子曰:朝闻道,夕死可矣。
——《论语·里仁》
上士闻道,勤而行之;中士闻道,若存若亡;下士闻道,大笑之。不笑不足以为道。
——《道德经·第四十一章》
周刊第7期中我说过自己的编程经历, 也提到目前主要写C代码. 我对C并不是很排斥, 因为它毕竟是我接触的第一门计算机语言, 印象不错. 但是我对于C++就很排斥, 因为它实在是 太 复 杂 了, 复杂到没有人敢说自己精通C++, 连IT公司都只能为C++划定一个范围, 程序员只能使用范围之内的C++. C++这个名字明明白白地表明了我对它的态度. 如果有人给我说, C++就是扩展的C, 我只想说, 艹. 如果我说艹艹, 那我指的是C#. 不过这是另外一个故事了.
我内心十分排斥C++, 遇到它就绕道走, 惹不起咱还躲不起么? 可生活有时就是这样, 你越是刻意回避, 越是会狭路相逢, 不得不硬着头皮会面. 最近的一个编程项目需要使用运算符重载. 查了一圈, 熟悉的语言中只有Fortran, C++, python支持. python做数值计算太慢, 就不考虑了. Fortran算是我的旧爱, 可以考虑, 但别人写的库代码压根就没有Fortran的份, 只有C++. 我倒是可以改写为Fortran的, 但是扪心自问了一下, 这样不累么? 算了, 改写成Fortran也学不到新东西, 还容易引入bug, 还是入C++的道吧. 能省则省.
这样看起来, 我虽然对语言有很强的倾向性, 却不是原教旨主义者, 一切都以实用为主. 我可以很排斥, 甚至讨厌某种语言, 比如python, english, 但我也不会有多坚持, 该用的时候还是会用起来. 这就是我常常自嘲的理想现实主义者. 如果遵照忠诚不绝对, 绝对不忠诚的说法, 那我妥妥该算作叛徒了.

以前, 这个脚本计算MM相互作用的时候支持两种组合规则, 分别对应AMBER规则和OPLSAA规则. 可有些力场却不使用任何组合规则, 原子之间的相互作用参数都是直接指定的. 虽然从优雅度上看, 这些力场都属于异端, 烧死最好. 但是不优雅的却可以很现实, 给出的结果可能更好. 毕竟, 大自然不是因为优雅而存在的, 优雅有时只是理论家的臆想或偷懒的理由. 所以我的脚本还是支持这种作法吧.
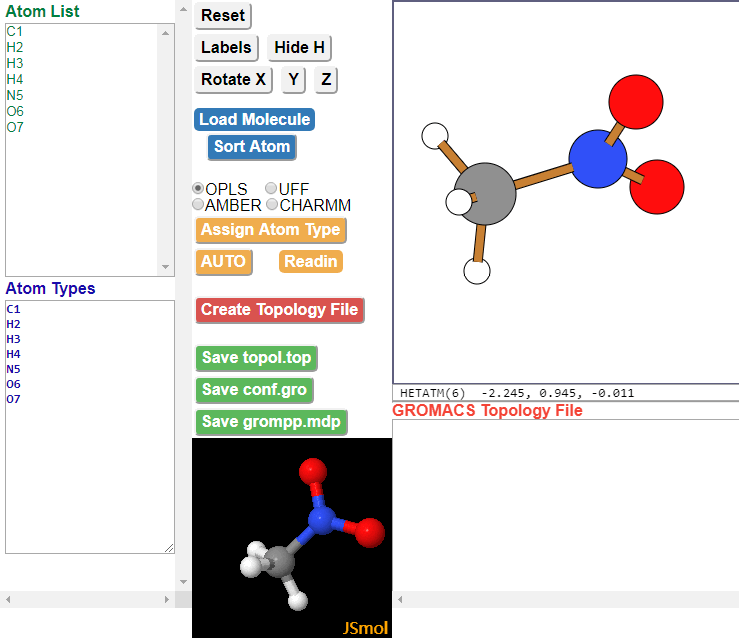
我一直想要把这个工具更新一下, 现在总算慢慢做起来了. 我决定让它支持AMBER, CHARMM, OPLSAA, UFF, MMFF94力场, 当然都是用于处理小分子的. 前几个都好弄, 最后一个有点麻烦. 好在像我在第2期中说的, Jmol已经有了一个MMFF94力场的实现, 所以我就先借助Jmol好了.
pymol的默认设置不用怎么魔改就能出效果不错的图. 有免费版的pymol, 但与正式版相比有不少差距. 试用版的pymol渲染图带有水印, 总让人觉得不爽, 就想着能不能想个办法把水印去了. 寻了一圈, 网上也没找到破解或去水印的方法. 当然, 如果你有授权文件, 或者用图片后处理去水印的方法, 就没有什么难度了. 既然没找到已有的解决方法, 那就试着自己做吧, 虽然这不是值得夸耀的事情, 事实证明也是如此. 最简单的方法, 将程序反汇编一下看看能不能找到点蛛丝马迹. 为此, 还专门学习了一下软件破解的知识, 毕竟以前还真没弄过这个. 试验了半天, 一无所获, 看来是超出了我目前的能力范围, 放弃了. 既然没法直接去掉水印, 那能不能在渲染的时候转个角度, 让水印出现在不需要的位置, 然后直接切除呢? 啊哈, 你很聪明, 可写程序的人也不傻, 他们早就想到有人可能会这么做, 所以图片中水印出现的位置是随机的, 每次渲染都不一样. 还是不行, 放弃了. 旋转水印不行, 那能不能把相机和分子转一转, 然后渲染呢? 试验证明还是不行, 水印是和相机关联在一起的, 并不是和屏幕关联在一起. 唉, 黔驴技穷了, 放弃吧.
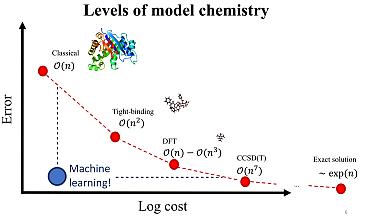
ANI神经网络势开发者的幻灯片, 可用于了解其背景和发展.
学艺术和建筑的, 经常要外出采风和写生. 做科研的也同样需要, 只不过换成了阅读文献和查看问题. 阅读别人的论文其实就是采风, 而尝试解决别人提出的问题, 就是写生了.
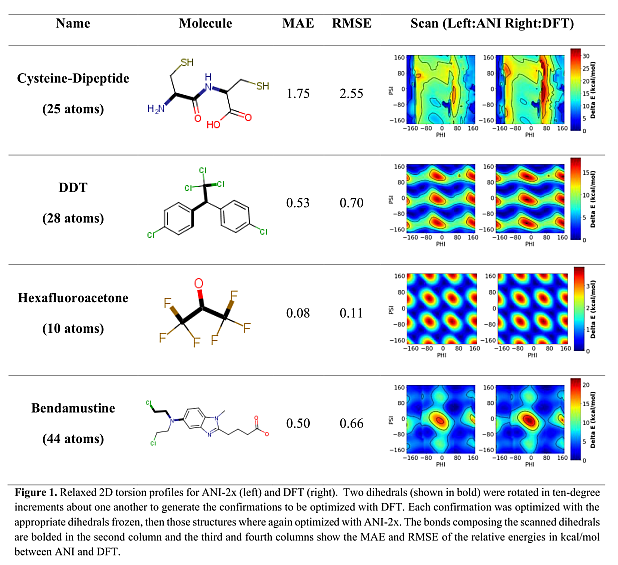
第2期中介绍过的ANI势, 到目前为止处理的元素已经扩展到H, C, N, O, F, Cl, S. 有了S, 就可以处理蛋白了. 但正式的参数网上没找到, 问了下作者, 原来文章还没正式出来, 等正式出来后, 参数就会发布到网上. 他们的下一篇文章应该会给出P的参数, 这样DNA也能处理了. 不过薛定谔早已经有了H, C, N, O, F, Cl, S, P的参数, 虽然不是免费的.
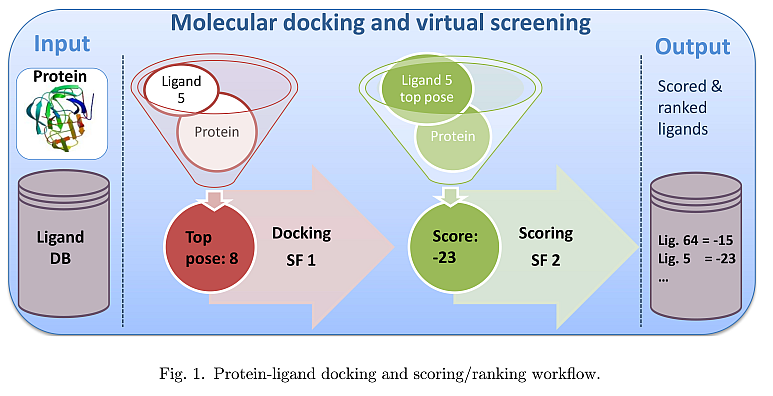
等集齐了ANI参数之后, 我想看看这个ANI势能到底能不能用来做对接, 方法可以参考这篇文章.
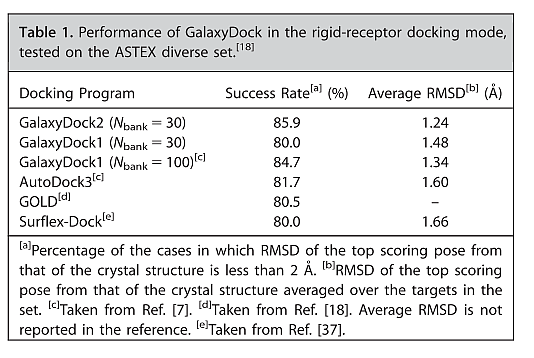
这是一篇对接程序的文章, 写对接程序的时候可以参考.
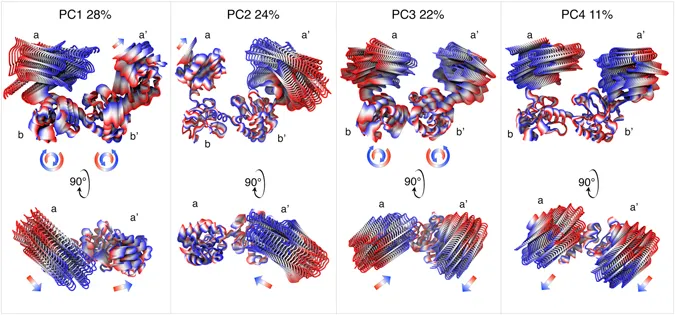
这篇论文的图都很漂亮, 其中的PCA图很多人都不知道怎么做, 先放在这里, 等我搞定了pymol之后就那它来试刀.
Gold dock6 sybyl, 特别是dock6有很多BUG的功能
Gold 据说评测分很高,但我没见过几篇文章。
我觉得gold打分函数不错,操作也简单. 所以通常不用复杂的设置,结果就可以
比较公认的准确度比较高的是薛定谔的gilde,还有一个是gold。免费的应该是auto dock vina吧,也有人说ledock准确度高,但是有人用100多个晶体数据做比对的时候发现结果并不怎么好(数据库名称不记得了……中科院一个人做的)一说igmdock也不错,但是结合口袋比较难弄,弄不好很容易出错。rostta对于对接软件测评的论文很少涉及到,因为比较难用,所以不知道准确度如何
网上有不少多肽对对接的服务器
你的设想可行, 但有两个问题不好解决: 1. Fe3O4的力场, 这是一种无机物, 很可能找不到合适的力场; 2. 模拟结晶过程可能需要很长长长长长长长长长长长的时间, 因为体系可能会过冷, 处于亚稳态很长时间. 这里的很长是相对MD的模拟时间来的, 微秒以上就算很长了, 实际中过冷液体至少可维持秒的时间
程序语言史记之JavaScript传.
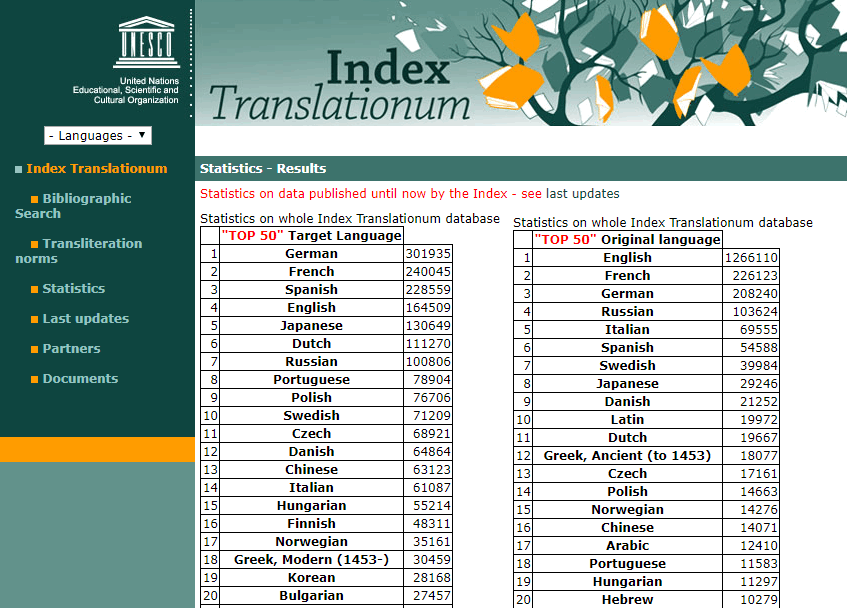
翻译目标语言中文是第13位,原创性处在第16位。

在语言网络中,中文还是边缘孤岛处境,区区一亿人使用的日文的中心性都比中文高,翻译量是中文的两倍。
本周刊记录我每周所读所思, 并自觉值得与大家分享的内容.
本周刊同步更新在我的网络日志 哲·科·文 和微信公众号 分子模拟之道.
如果你觉得我的分享对你有益, 不妨将它推荐给你认识的人.
如果你也认同分享的理念, 欢迎投稿或推荐自己的内容. 请关注微信公众号后台留言, 或加入QQ群联系.