
我年纪也不小了, 但在父亲面前, 当然还算是孩子. 回了故乡, 长辈见了, 大多也是叫我的小名. 我常记起父亲给我们讲他年轻时候的事, 说起那时生活的艰难, 说起他年轻时候的无畏与力气, 修水库, 推小车, 一顿吃几斤饺子. 说到这些, 他有时就看不上我们这代, 觉得我们没吃过苦, 没受过累, 条件比他们那时好了很多, 可力气却没有, 干不得活.
就是在这样的情形下, 我至今还记得, 他有一次问了一个问题: 为什么他们那个时候的人受那么多苦, 出那么大力, 没黑没夜地劳作, 可还是那么穷, 穷得叮当响; 而现在的人受不得苦, 出不得力, 生活条件却好很多呢?
是啊. 他们那时辛苦劳作却得不到好的生活, 究竟为什么呢? 如果人类的劳动是无差别的, 对应的价值也是统一的, 那么自然付出劳动就有收获, 多劳多得, 无法不劳而获.
现实自然不是这样. 一个战乱之地的农民辛苦劳作, 一个中国的农民辛苦劳作, 一个美国的农民辛苦劳作, 即便他们付出同样的劳动, 所得也大不同. 同样的辛苦劳作, 在有些地方只够生存, 在有些地方可以生活, 在有些地方可以享受一点生活. 这是为什么呢?
这其中的差距与问题, 父亲大约只是有些感觉到, 没有能力深入思考. 他最后告诉我的是, 因为他们那个时候国家穷, 没有钱, 而现在国家富了, 有钱了. 这应该是他自然的想法吧. 因为他觉得国家就和一个大家庭一样, 整个家庭穷, 没有钱, 那家里的每个人就要辛苦点, 否则就维持不下去了. 可如果整个家庭有钱, 家里的每个人不用那么辛苦也可以生活下去.
父亲只读过几年小学, 只有他朴素的想法, 不会继续追问下去: 为什么国家以前穷, 现在没有那么穷了, 穷到不穷的转变是怎么发生的?
这确是个大问题, 值得每个人思考.
【李继存】分子动力学模拟视频主题讨论
【时 间】2020-05-24 周日 晚 20:00-21:00
【参 与】加入QQ群: 132266540GROMACS/AMBER中文组), 按时上线加入QQ电话/群视频
【内 容】讨论合作事务, 确定待录制视频的主题和主讲人.
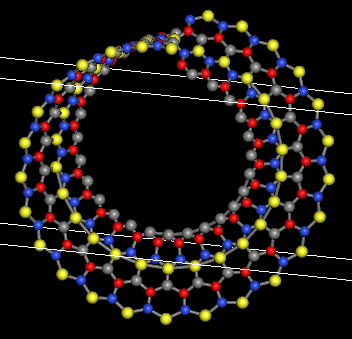
我就知道, 该来的总是要来, 就像那个墨菲定律. 我不把该做的功能做上去, 总有一天会有人找上來. 这个石墨烯建模工具本来挺简单的, 我眼看着它越来越复杂. 你要这个功能, 我要那个功能, 功能越添越多, 程序越来越繁, 最后大致肥胖而死, 重新托生, 然后继续轮回.
现在我们要加一个将二维石墨烯片卷曲的功能, 就像将一张纸弯曲成其他形状一样. 处理方法另文细说吧, 这里只说结果: 使用参数方程, 沿XZ方向扭曲.
吹起海螺, 666

我的小心脏, 跳跳跳

卷个戒指送给你

一颗爱心 5.21

千旋万转也只有一面
数据库中的蛋白都是有二级结构的, 如果要得到没有固定二级结构的蛋白模型, 可以试试聚合物建模中常用的自回避行走模型. 使用这种模型生成的无规蛋白更合理一些. 如果使用随机行走模型建模, 那大概率原子间会有重叠, 导致最后无法优化.
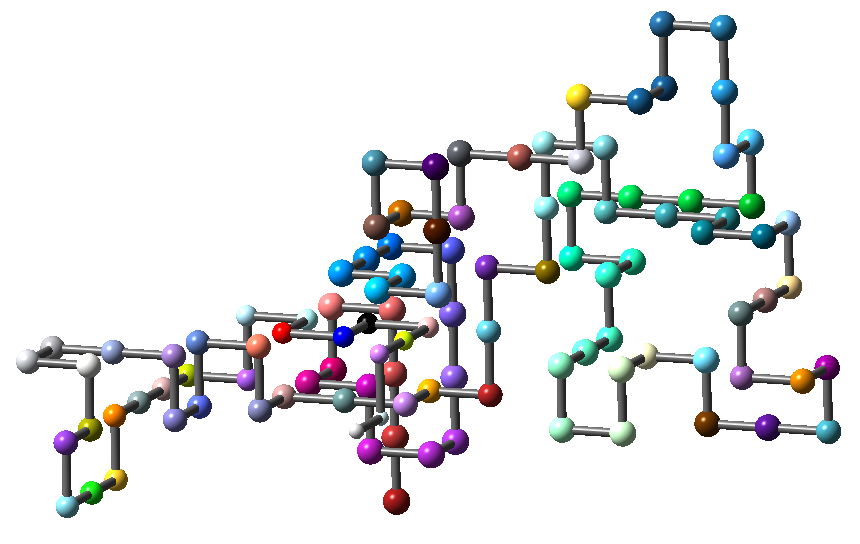
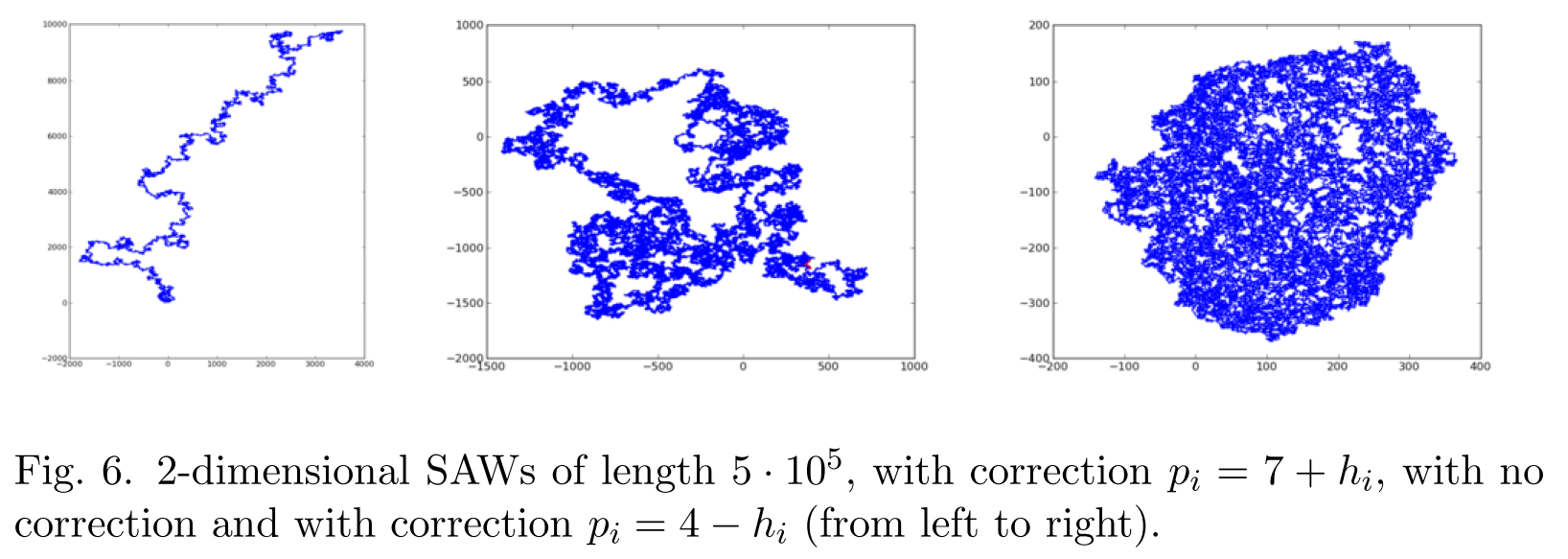
普通的自回避行走模型运行到一定程度会锁死, 无法得到任意长度的链. 这个python代码可以生成任意长度的自回避行走链, 并具有可扩展性, 还可以使用参数控制链的密实程度. 所得结果可以作为聚合物或粗粒化模型的初始构型.
学艺术和建筑的, 经常要外出采风和写生. 做科研的也同样需要, 只不过换成了阅读文献和查看问题. 阅读别人的论文其实就是采风, 而尝试解决别人提出的问题, 就是写生了.
可扩展性自回避行走模型的论文, 给出了数学细节和一些例子.
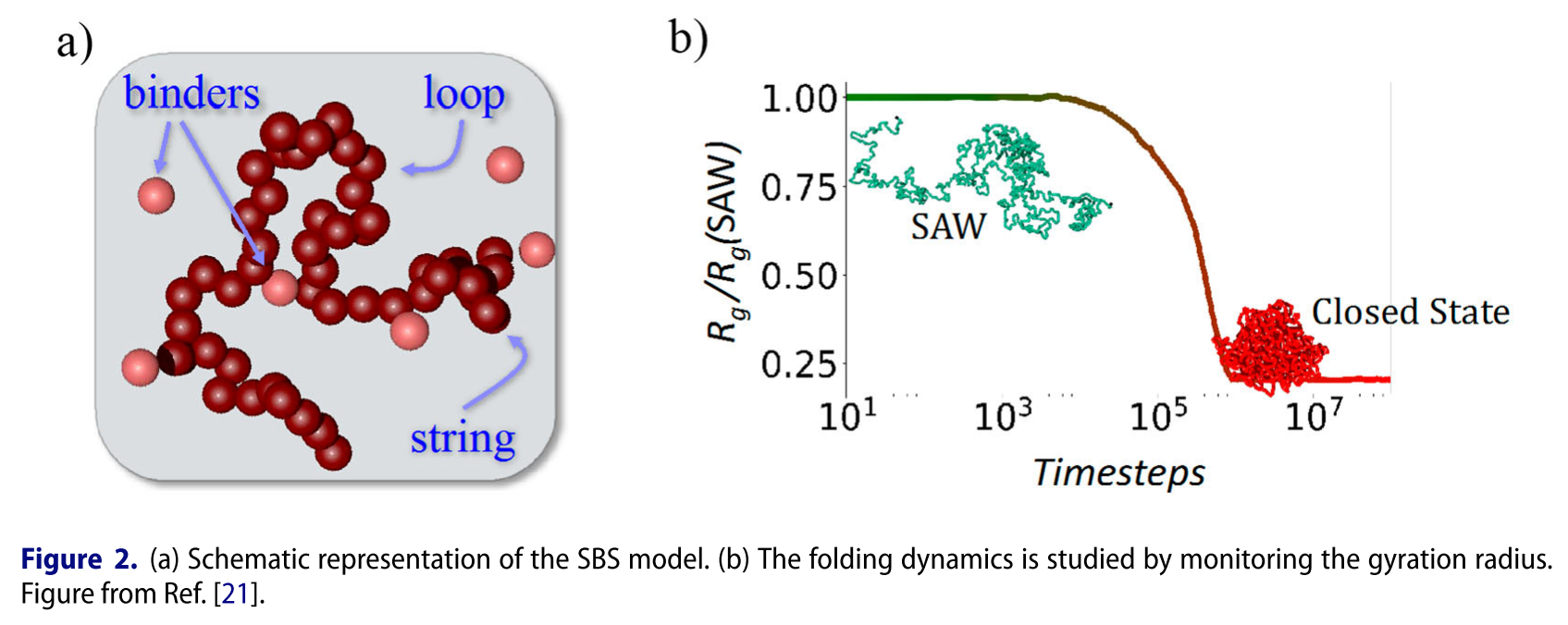
聚合物建模的文章, 基于粗粒化的势能函数, 运行MD, 得到聚合物的构型. 实现还算方便, 可以作为GROMACS的一个示例教程.
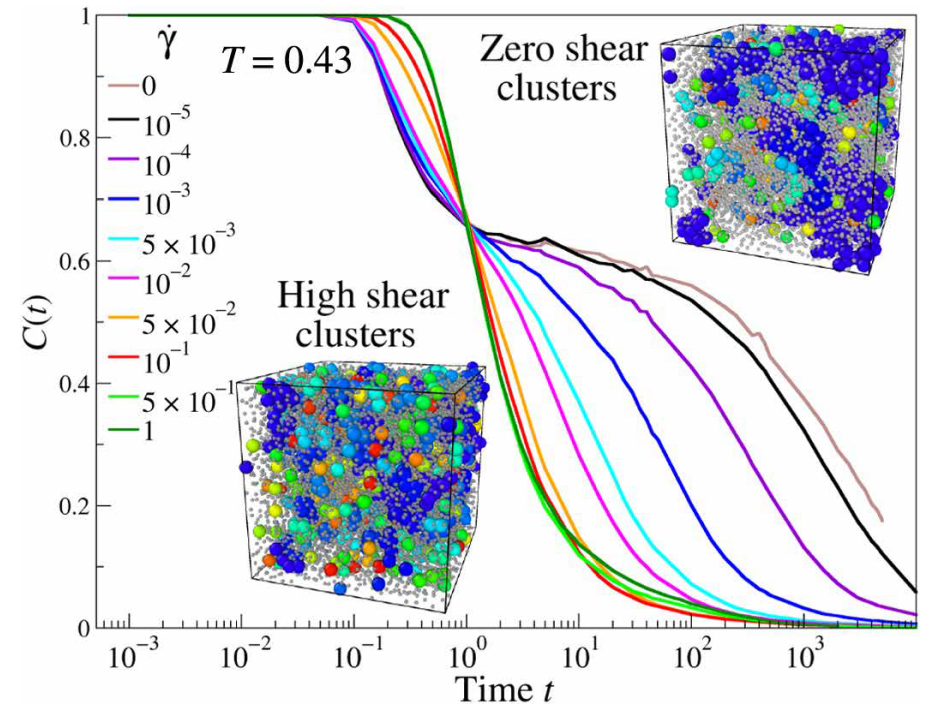
利用粗粒化模型研究聚合物剪切的文章. 知乎有一篇解读. 与前一篇结合起来, 或许可以灌不少水.

讨论聚合物缠绕的文章, 给出了数学化的定义, 知乎有人评论.
这个题图有点像我印象中的染色体形状. 难道染色体也是缠绕的?
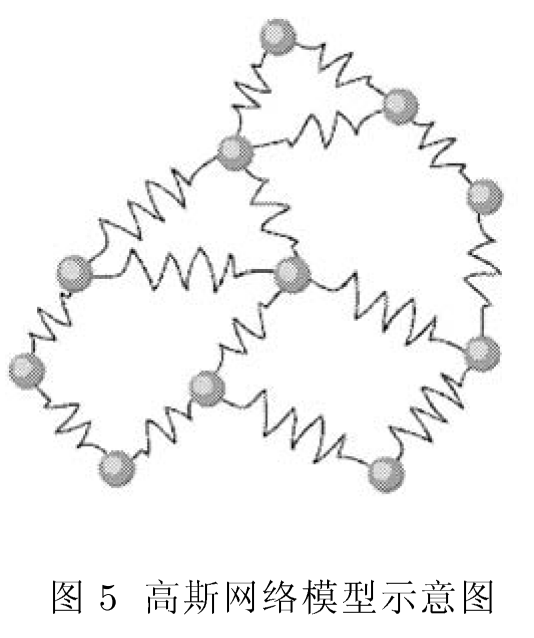
比较老的中文文献了, 重点讨论蛋白质模拟的粗粒化方法, 对理解粗粒化有帮助, 虽然那个时候这个名称还不流行. 这些方法同样适用于聚合物.
全原子力场的拟合本来就是个见仁见智的问题,没有什么绝对准确十全十美的力场。所有力场参数的产生都是去拟合某个参考数据,可以去拟合能量数据,或者受力,或者实验数据,等等。所以得到的力场参数都是在某些方面表现得很好,但在某些方面表现得不好。有的力场甚至在某些温度下表现好,某些温度下表现不好,这都是很正常的。
这是因为,拟合全原子力场,和量子化学从头计算相比,本身就是一个粗粒化(coarse grain)的过程,把电子自由度去掉了,粗粒化就意味着信息的丢失,但想定量分析哪些信息丢失是一个很复杂的问题。
你说的那些脚本(比如Amber, CHARMM里的脚本)可能是一些通用的全原子力场生成小分子力场的脚本,因为人们不可能每拿到一个新分子就去拟合一个力场,那样太费力了,像Amber, CHARMM这样的通用力场在定性方面能够满足大多数人的需要,所以就被直接拿来用。如果想追求定量分析的化力场参数还是要精确拟合得到。
现在不少文献都不严谨的,但大多数文献如果自己拟合了力场是会提到自己拟合力场采用的软件或者算法(一般在参考文献里),拟合得到的力场参数也会在supporting information里给出。
力场参数的拟合是很复杂的问题,特别是涉及几十个参数同时拟合,在技术上很大程度上取决于你的算法,对于新手或者组里没有开发力场经验的筒子,一般不建议自己去拟合力场。
二面角扫描再拟合参数,之前我就至少看到过两篇,一篇是12年的jctc,一篇15年的jacs,高水平文章是很严谨的。
在线程序或者脚本得到的itp基本都要自己填一些参数的吧,比如最基本的电荷肯定不能用他自己生成的,自己改下呗,其他参数根据自己需要去改,拟合参数也可以根据研究内容拟合对其影响较大的一些参数,不关键的就用原来的。
我现在就在做力场拟合, 具体过程知道的多些. 单对小分子力场来说, 使用量化程序计算能量或力拟合力场参数工作量不大, 可行. 要是拟合实验数据的话, 就麻烦得很. 完全基于量化的力场拟合, 现在关键的问题是vdw参数, 没有通用的方案可以获取vdw参数, 其他都好办些. 电荷可以使用resp的或其他的, 分子内参数可以拟合, 都很说得通. 所有目前很多人做力场都不是完全基于量化的, 大部分是在某一力场, 如charmm, oplsaa等的基础上添加或修正一些参数, 这样就无需重新拟合vdw或电荷. 如果你的原子类型确定了, vdw参数确定了, 电荷确定了, 扫描构型, 无论是键长, 键角或是二面角, 然后来拟合其参数, 还是很容易做的. 其中要注意的是, 不同构型之间这些参数间可能有耦合, 需要综合处理, 但并不麻烦.
见 图文专辑 分子模拟周刊
本周刊记录我每周所读所思, 并自觉值得与大家分享的内容.
本周刊同步更新在我的网络日志 哲·科·文 和微信公众号 分子模拟之道.
如果你觉得我的分享对你有益, 不妨将它推荐给你认识的人.
如果你也认同分享的理念, 欢迎投稿或推荐自己的内容. 请关注微信公众号后台留言, 或加入QQ群联系.
