
搬到一个新的公寓, 租金不少, 风景不错. 透过阳台的落地窗, 便可以看到路对面一条有名字的河, 查尔斯河. 这条河在公寓前面扭了扭身子, 便拱出一片浅水区, 近乎湿地, 岸边很适合散步和垂钓. 河边上生长着各样的树, 还有很多我不知名字的刺藤, 放肆地侵占.
亏了是住在这个地方, 隔离在家期间没太耽误外出放风, 门前也没少了钓鱼的人, 时多时少. 孩子是不好一直关在家里的, 只得不日带她出去沿河边巡逻, 到岸边高地巡山, 这些都成为了她探险的一部分. 因为对她来说, 确实也有险的地方, 比如掉到树根缠绕的陷阱中, 穿梭时被刺藤划伤, 遇到不常见的蛇啊.
我喜欢站在窗前观看朝阳夕辉映照下的河边树林, 一切仿佛沉睡在无边的静谧中, 让人觉得安详, 暂时忘了现实的沉重与喧嚣.
我喜欢岸边的那片松林, 那里杂草不多, 地面落满枯黄的松针, 踩上去软软的, 闻起来有着松树特有的香气, 风吹过的时候, 更能听到微微的涛声, 虽然比不上原始松林的怒吼. 每当在那里听见风起松涛, 我常遐想, 隐到最北方的松林, 不问时事, 白日读些文字, 然后每夜每夜地听那些真正松林的涛声, 就想着海.
而这些, 我身边这个小小的人儿暂时都还没有办法理解, 虽然她感情丰沛, 有着更加细腻的心思.
<video controls="" preload="none" poster="https://jerkwin.github.io/pic/weekly/21_mp4.jpg"><source src="https://jerkwin.github.io/pic/weekly/21_0.mp4" type="video/mp4"></video>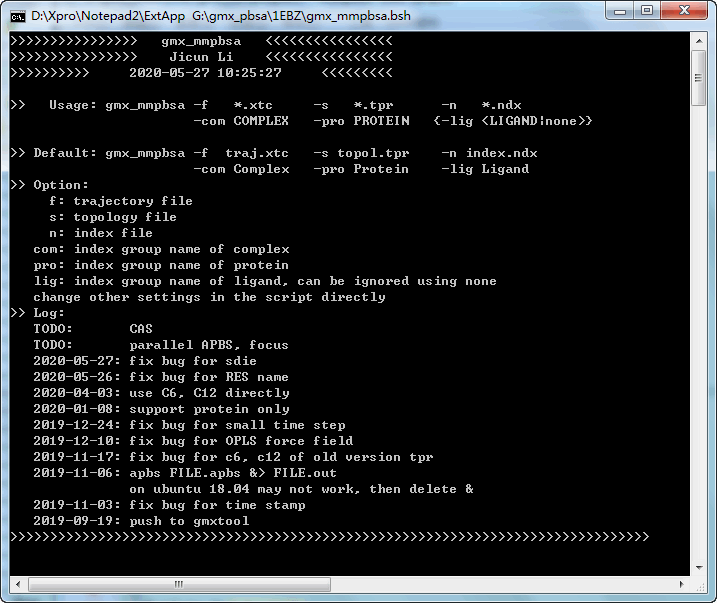
这个计算MMPBSA的脚本已经发布很长时间了, 也不时有人在用. 既然有人用, 那就会发现问题. 最近又有人发现脚本存在严重问题, 所以就集中修订了一次, 把前面累积的一些问题都解决了, 并改写了脚本运行方式, 以方便使用. 如果你需要计算MMPBSA, 请用最新版本. 如果发现问题, 请告知以便更新修正.
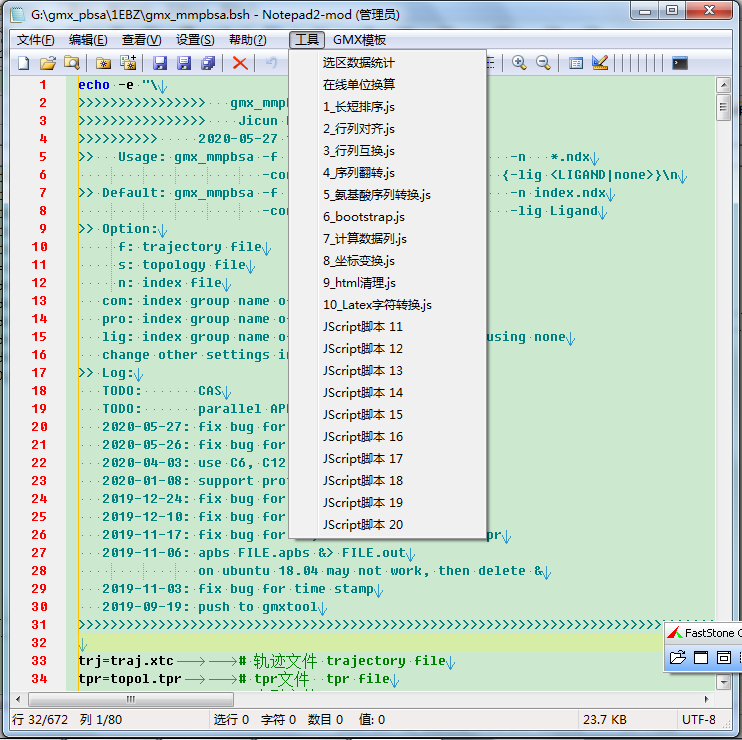
以前发布过一个Notepad2的修订版, 是我基于原始代码编译的, 但增加了很多自己需要的功能. 最近又给它增加了插件功能, 也就是可以调用外部的脚本. 目前仅支持windows的JScript脚本. 虽然也可以很容易地支持VBscript, 但我觉得这种脚本适用面太窄, 就先不考虑了. JScript是微软自己实现的JavaScript, 已经废弃, 不再更新, 但基本的语法和js一致, 处理简单的文本已经够用了. 更可贵的是, JScript脚本在windows下可以直接运行, 而且还可以借助系统函数实现一些js无法实现的事情, 比如操作剪切板, 显示对话框等. 此外, 这种脚本所用的代码很容易改写成浏览器版的js代码, 从而做成在线工具.
继续考虑的话, 下一步看来是让这个插件系统支持更多的脚本语言, 比如最常用的bash, python. 这样可以大大提高Notepad2的功能, 但不用改变核心代码.
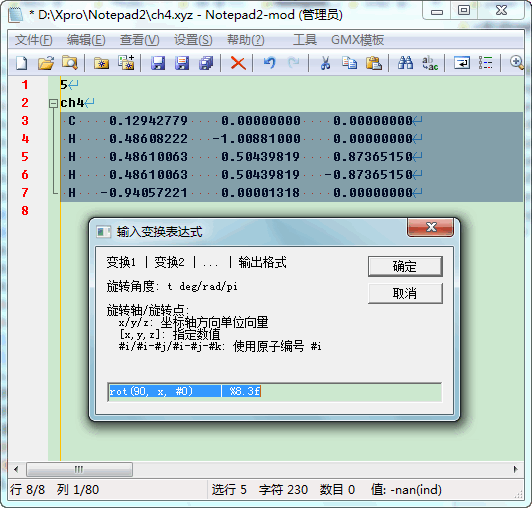
前些日子整理过VMD坐标变换的手册文档, 并给了一篇建模示例教程. VMD对分子坐标的变换功能确实很强大, 但使用过程中总觉得有点不方便. 有时我只是想简单地进行下坐标变换, 如果沿某个轴对齐, 绕某个轴旋转, 却也要打开VMD, 写好几句tcl代码, 然后再保存才能得到坐标. 这种模式对于简单的变换操作来说实在有些过于繁琐, 所以我就比着VMD的坐标操作方法, 写了一个JScript脚本, 可以直接对选中坐标进行各种变换, 得到变换后的坐标, 使用方法方便了不少.
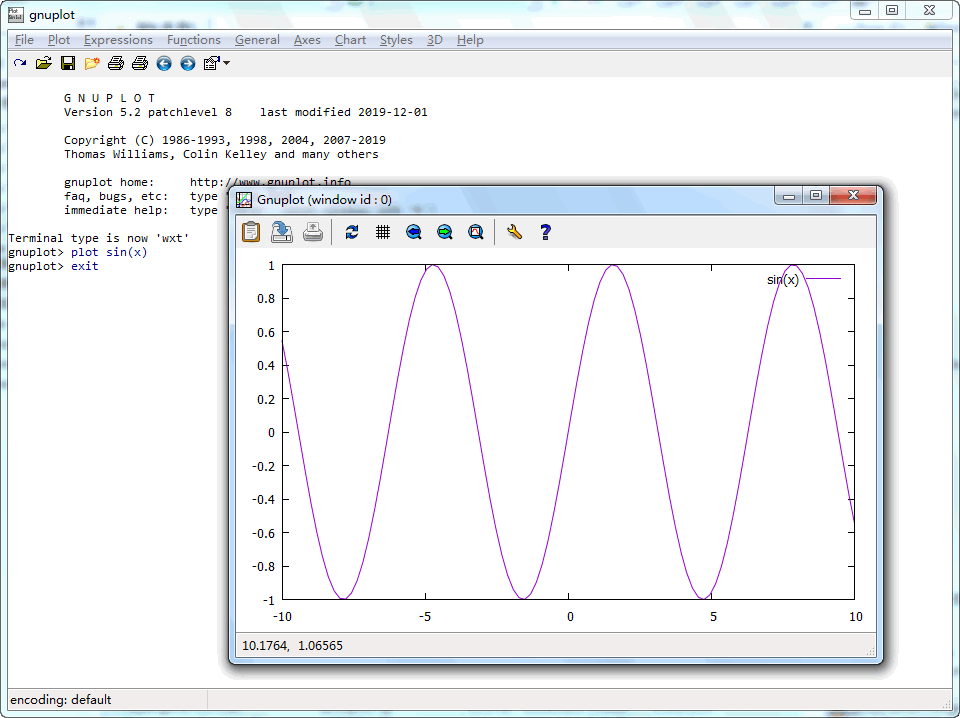
把gnuplot更新到最新5.2.8版, 然后整理了一下自己所用的相关脚本, 初始设置. 这样操作模式基本就定下来了, 以后也不用大改了. 安装配置过程有待整理发布.
学艺术和建筑的, 经常要外出采风和写生. 做科研的也同样需要, 只不过换成了阅读文献和查看问题. 阅读别人的论文其实就是采风, 而尝试解决别人提出的问题, 就是写生了.
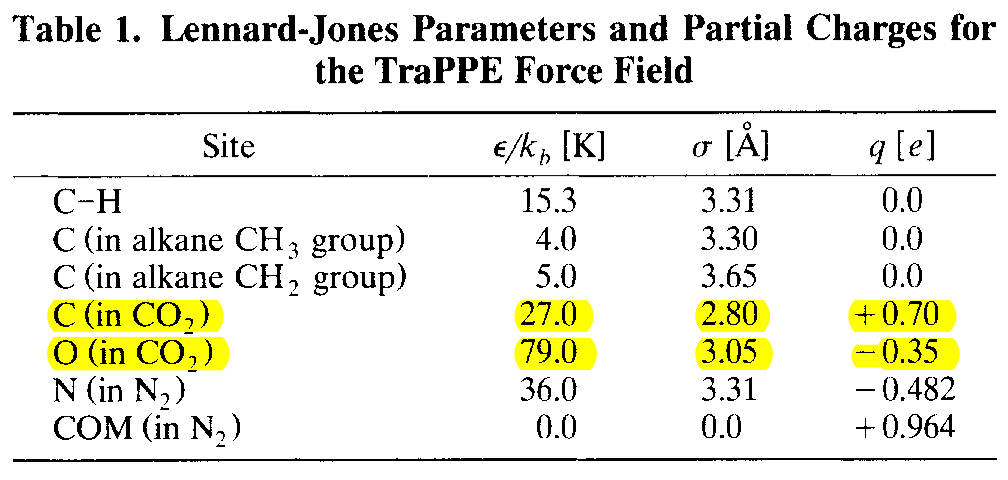
最近需要用到二氧化碳的力场, 就查看了下文献上用的比较多的CO2力场. 这篇文献是TraPPE力场的原始文章, 给出了CO2的力场参数. 值得注意的是, 文章使用的MC方法, 所以CO2是刚性的, 如果直接在MD中使用这个模型, 还需要采用一些特殊的方法来确保模型的刚性.
这篇文章提到开发了一个TraPPE力场的数据库, 可以查看各种参数. 但网站年久失修, 已经无法打开. 具体位置待考.
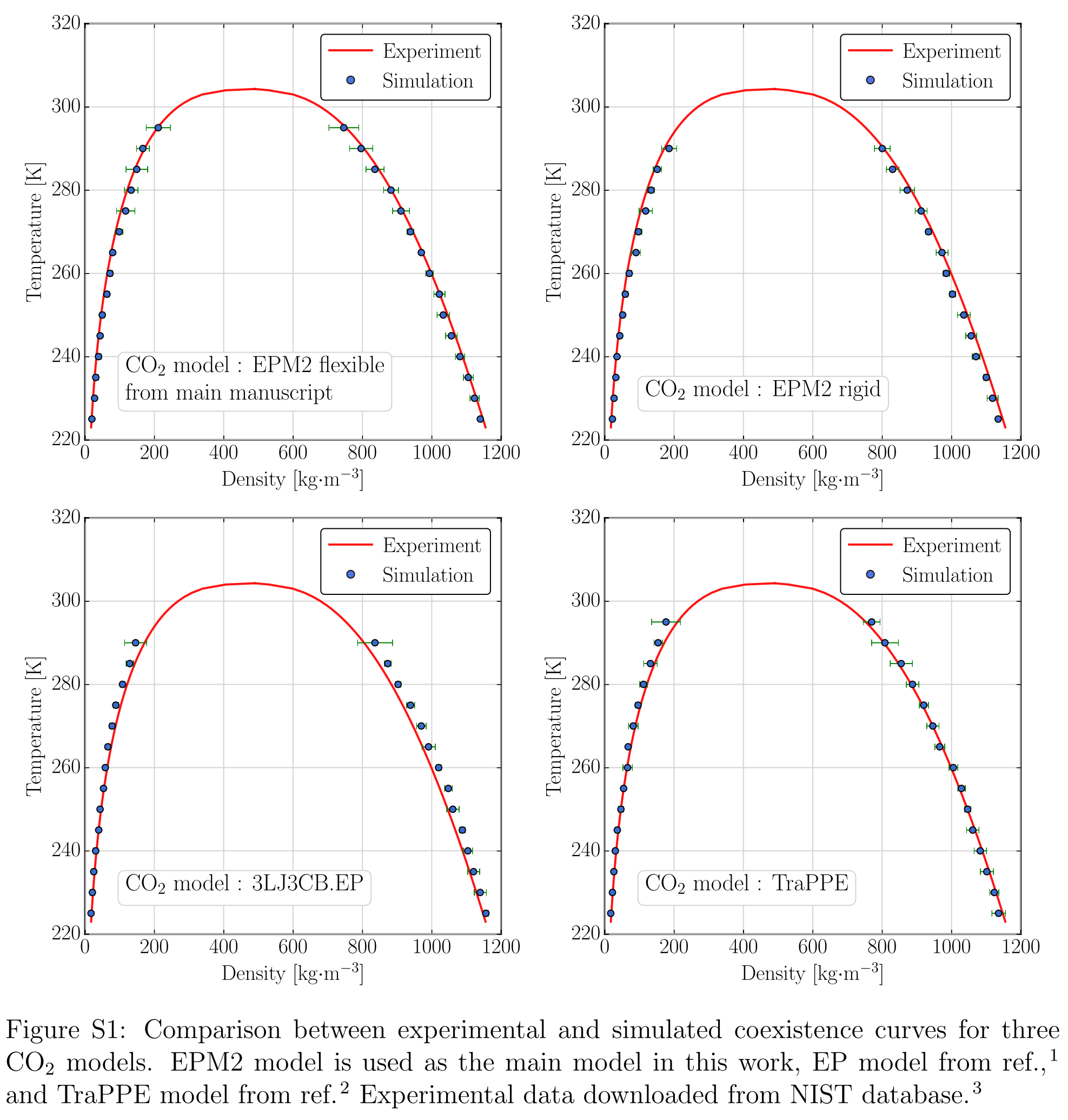
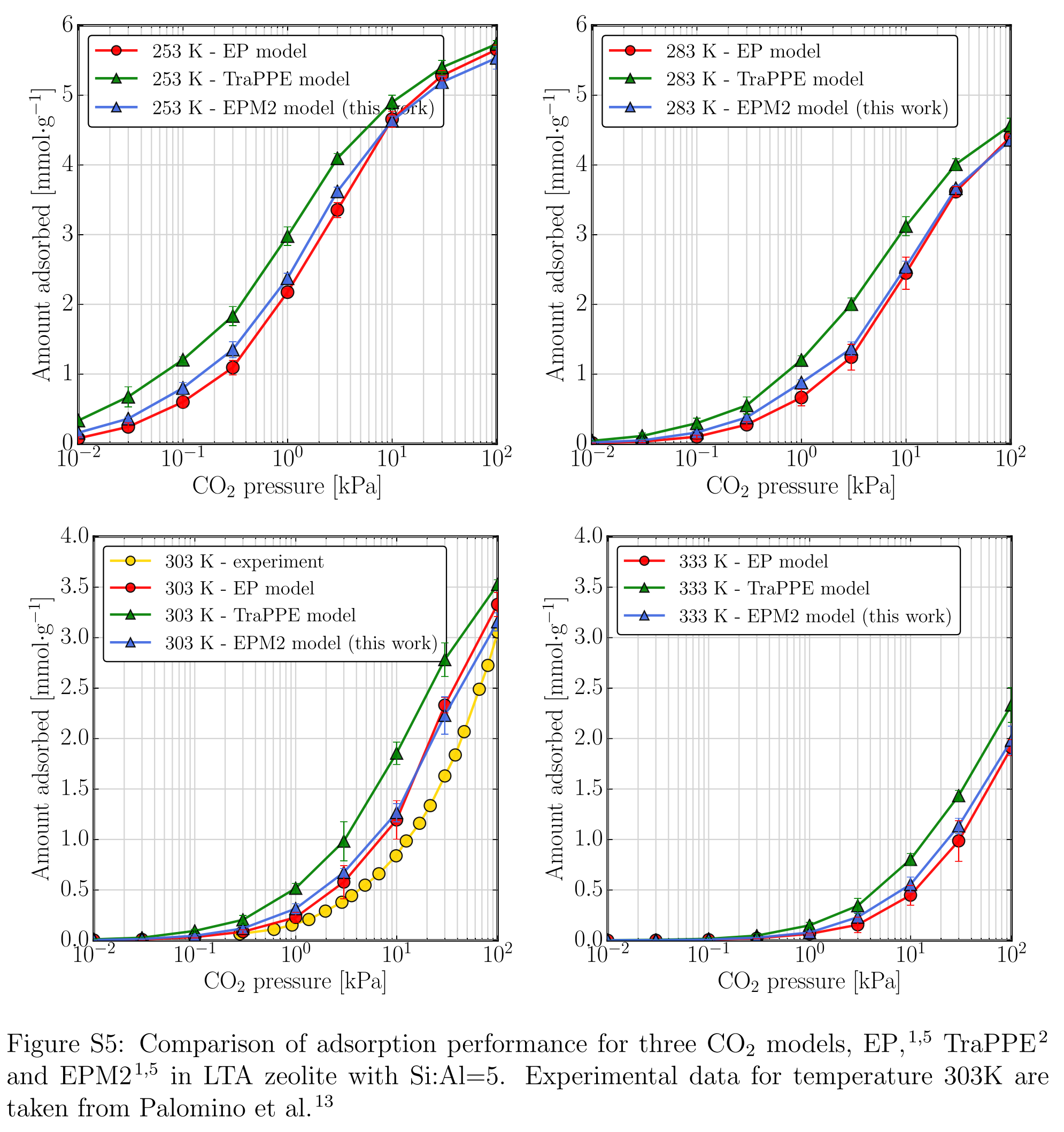
这篇文章算了几个气体小分子在沸石上的吸附, 其中CO2用了多种力场模型, TraPPE, EPM2刚性, EPM2柔性, EP. 如果要做类似研究, 可以根据这篇文章的结果合理化一下自己所用的力场.
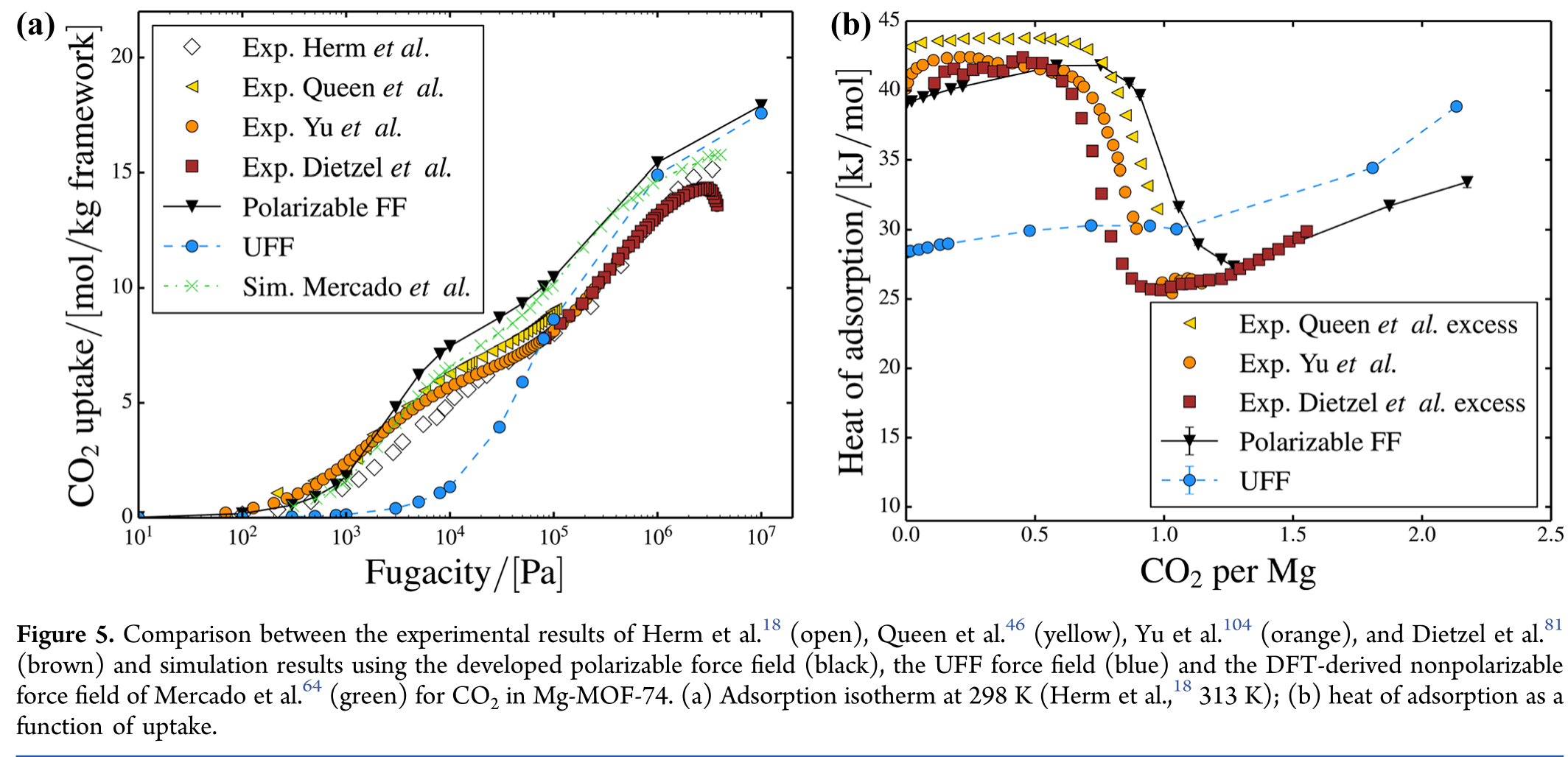
这篇算的是CO2和CH4在MOF上的吸附, 用了极化力场. 如果喜欢使用新力场, 可以试试. 文章结果表明UFF力场不够好. 这也很正常, 因为UFF力场很粗略, 是实在没有力场可用时候的备用. 太通用的力场你不能要求太多.
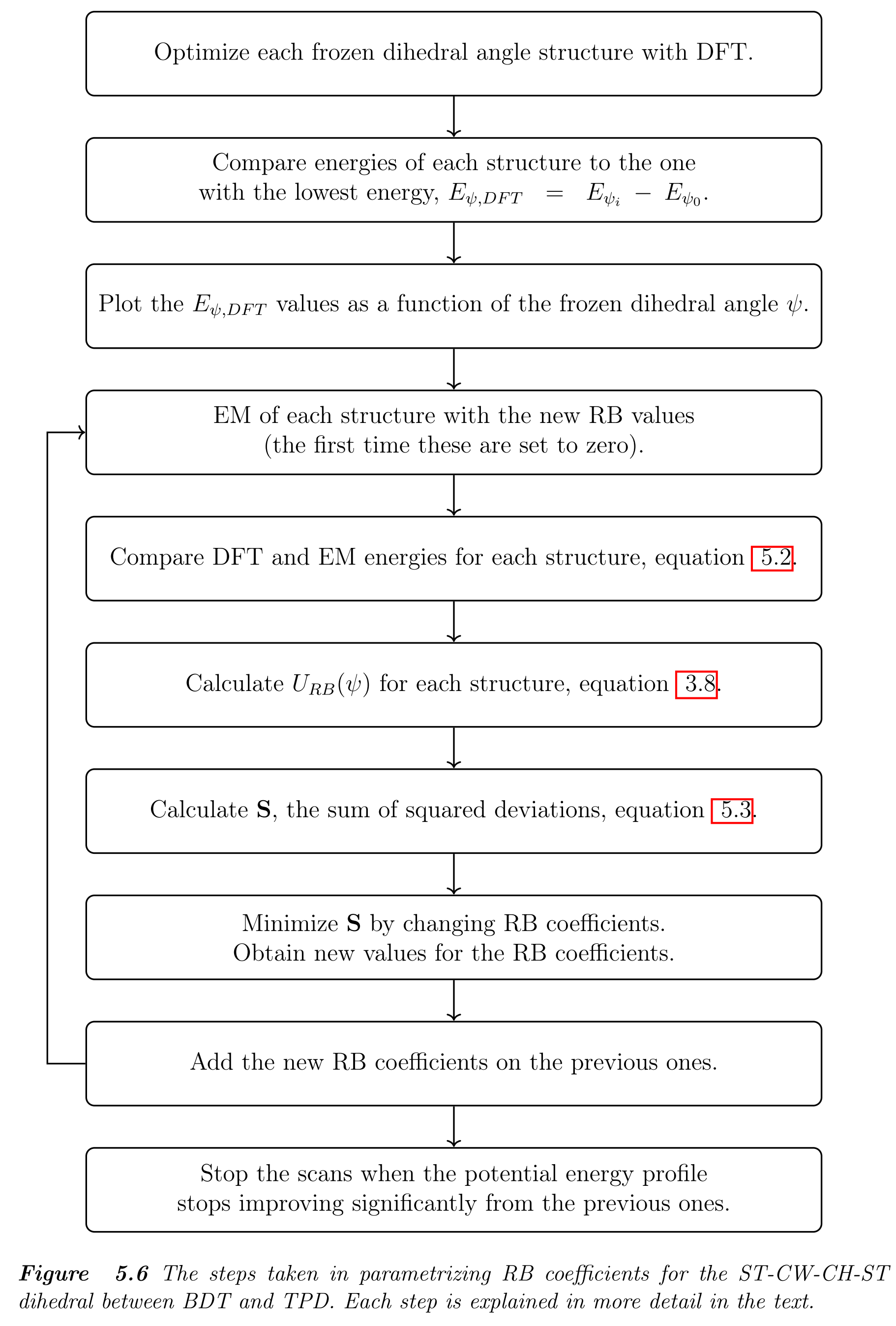
这是篇硕士毕业论文, 质量一般. 其中有些MD的基础知识可以读读, 复杂体系简化处理方法, 聚合物建模也可以参考. 对想要自己拟合力场参数, 特别是二面角参数的人来说, 论文中给出了详细的流程, 这部分最有价值.
计算氢键需要很大的内存, 体系太大的话, 内存就会不够, 这可能是你得到段错误的原因, 和skip无关, 因为对每帧都需要计算, 使用的内存每帧都是一样的. 如果可以的话, 使用gmx select选出你要计算的组, 尽量小, 这样就可以避免内存的问题. 比如计算蛋白和水的氢键, 水只需要选择距蛋白一定范围内的即可, 远处的不可能与蛋白形成氢键, 不需要考虑.
固体的话, 常温常压下不好验证, 因为模拟的时候基本不动. 要根据你要研究的性质来验证, 比如你研究力学性质, 你就要验证弹性模量, 研究界面性质, 就验证接触角, 研究融化, 就验证熔点. 通用一些的, 是验证密度, rdf, 配位数, 键的取向等.
你读过石墨烯的教程么? 里面说的很明白:
如果使用柔性的模型, 在模拟过程中石墨烯的结构会发生变化, 这就需要能够描述石墨烯变形的力场参数. 在这种情况下, 模拟时还可以有两种处理方式. 一种是将石墨烯视为孤立的分子, 不考虑其周期性, 这样在模拟时边界碳原子由于缺少约束, 可能会卷曲. 如果发生了卷曲, 可以通过固定一些边界碳原子来解决. 另一种处理方式是将石墨烯视为具有周期性结构的无限体系, 这样就不存在边界碳原子, 也就不会发生石墨烯卷曲的问题.
chimerea可以算体积, 还有个fortran程序也可以, Computer Physics Communications 165 (2005) 59–96, 但这些都需要定义原子半径.
基于量化的方法对小分子也可以, 但大分子就比较困难.
动力学半径这个概念是个实验上的概念, 没有简单的微观对应计算方法, 很多人是经验的估计. 两种方法: 1. 计算分子大小, 然后根据它们的动力学半径数据做回归, 然后用到未知分子上; 2. 基于电子密度面计算, jpca上有篇文章, 可以参考.
见 图文专辑 分子模拟周刊
本周刊记录我每周所读所思, 并自觉值得与大家分享的内容.
本周刊同步更新在我的网络日志 哲·科·文 和微信公众号 分子模拟之道.
如果你觉得我的分享对你有益, 不妨将它推荐给你认识的人.
如果你也认同分享的理念, 欢迎投稿或推荐自己的内容. 请关注微信公众号后台留言, 或加入QQ群联系.
