
C诡异离奇,缺陷重重,却获得了巨大的成功。
——Dennis Ritchie
换了工作之后, 编程的主打语言变成了C, 暂时不用FORTRAN了.
回想起来, C是我接触的第一门计算机语言. 这似乎是20年前我上大学时的惯例. 大多数理工科非计算机专业的学生所学的计算机语言都是C, 而所用的教材就是绿皮本的谭浩强, 虽然这本书现在被喷得厉害. 当年的我, 喜欢C语言的简洁, 类数学性, 也有一些绕脑的地方, 可以让你深入思考. 不过, 当年我也只是学了些简单的语法, 会写个hello world, 指针文件这些不知所云. 这样的水平自然不会想到, 也不可能将学到的C用于平时的学习.
读研之后, 导师写FORTRAN的, 我自然也开始学FORTRAN, 写FORTRAN. 入门书是谭浩强的FORTRAN 77, 彭国伦的FORTRAN 95. 自那之后, 就是我的FORTRAN时代了, 基本上各种东西都用FORTRAN写过, 无论是项目需要的, 还是平时需要的.
我的FORTRAN时代持续了15年后暂时终结, C时代重新开始了. 这次看的书则是 K&R (Kernighan and Ritchie)的 C程序设计语言. 这本书基本是C语言的圣经, 因为R是C语言之父, 而hello world也是自这本书开始的. 读到这本书的时候, 我才了解到K&R中的K也是AWK中的K, 也才明白了为什么AWK和C长得那么像, 像得让我觉得AWK就是脚本化的升级版C.
这本C, 我看起来没困难, 除了 指针. 我耗尽力气, 也还未尽掌握. 看来脑子还要绕段时间.
聪明如你, 一定知道下面这些变量的含义吧.
char **argv
int (*daytab)[13]
int *daytab[13]
void *comp()
void (*comp)()
char (*(*x())[])()
char (*(*x[3])())[5]
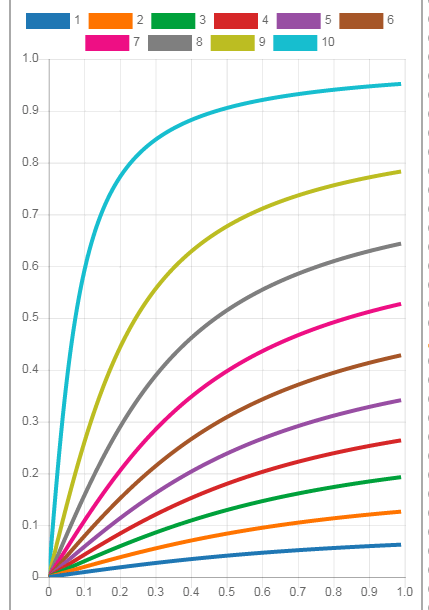
将自己所用的gnuplot配置脚本重新整理了一下, 其中的调色板也增添了几个. 如果你厌倦了某些软件自带的调色板, 可以到这里找找看有没有喜欢的.
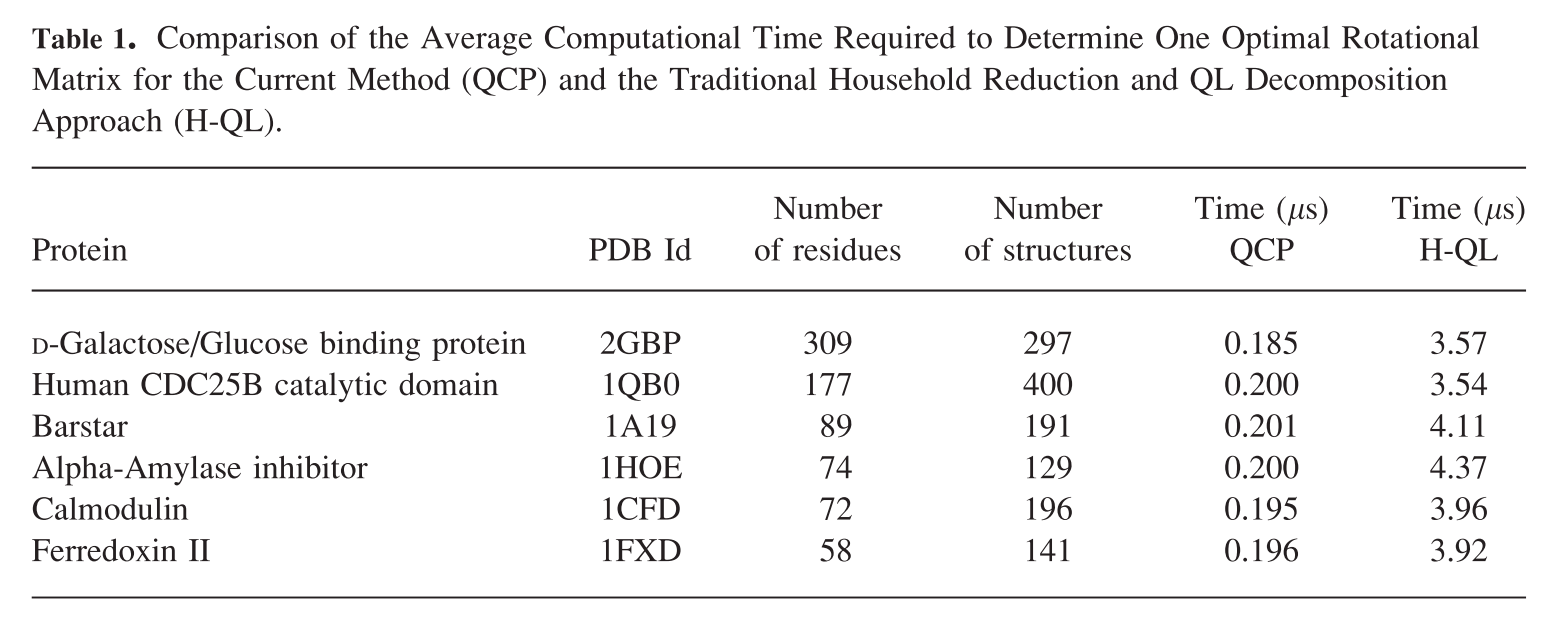
据说是计算RMSD的各种方法中最快的. 如果你有超大或超多的蛋白需要计算, 可以一试.
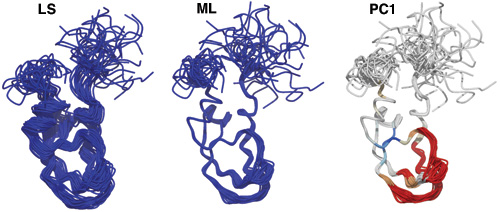
通常的构型叠合基于RMSD最小化, 这个程序使用的方法则不同, 是基于概率的. 对有些情况, 得到的结果可能更好.

嗯, 按这三本书的标题做, 你也能成为专家, 或者砖家.

看到不少人说这个机器翻译不错, 可以考虑加入到我的机器翻译脚本中.
学艺术和建筑的, 经常要外出采风和写生. 做科研的也同样需要, 只不过换成了阅读文献和查看问题. 阅读别人的论文其实就是采风, 而尝试解决别人提出的问题, 就是写生了.
最近用到了MC方法, 这篇短的综述对MC的介绍很好.
这篇力场参数优化的文章用到了MC, 讨论了具体的细节, 实现时可以参考.
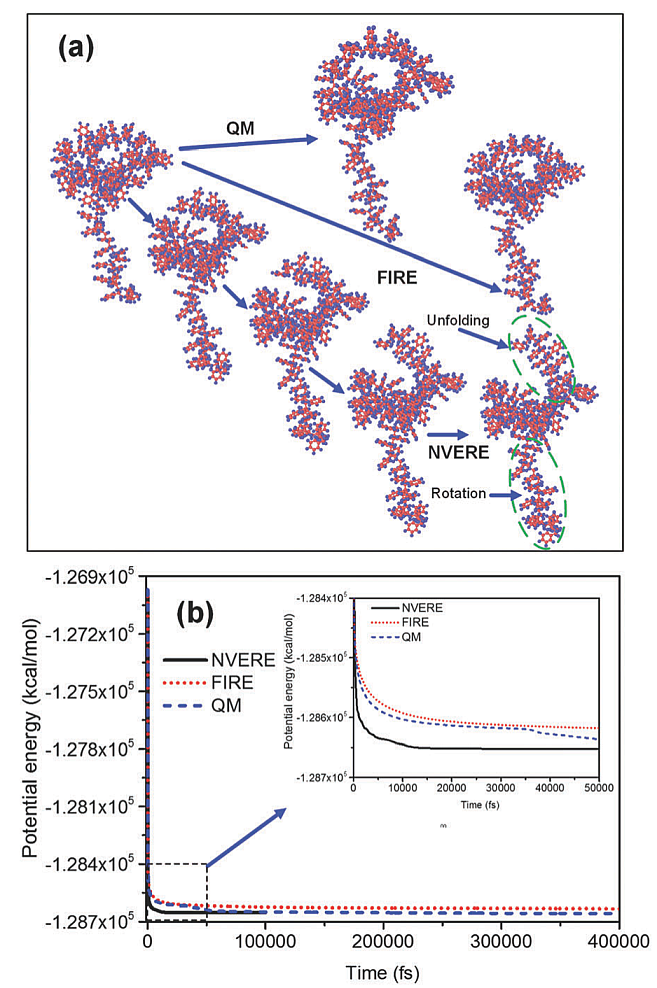
查FIRE优化方法的时候, 顺便看到了NVERE优化方法. 其作者宣称效果好于FIRE. 我没有测试, 持保留意见. 多嘴一句, 很多优化算法的文章比较时没有标准, 田忌赛马, 号称自己的方法又快又好.
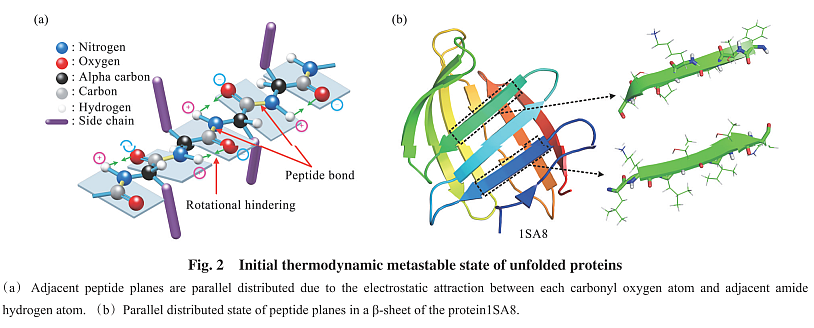
NVERE方法作者的论文, 分析蛋白折叠的机制, 有启发.
对应的英文文章 Physical Folding Codes for Proteins 只有预印本, 而且多出很多内容, 值得一看.
 (吕康杰提供)
(吕康杰提供)
那个参数的含义我说下吧, 但都是理论上的, 没有实际测试经验, 仅供参考.
运行并行程序的时候, 同一个任务要分配的很多不同的处理器上去运行, 每个处理器计算完成后再把结果汇集起来, 进行处理, 继续下一步, 理想情况下, 如果可以把任务平均分配到每个处理器, 每个处理器完成计算的时间相同, 这样所有的处理器同时开始运算, 同时结束运算, 效率最高, 因为不会有处理器处于空闲状态, 不要等待其他处理器, 这种理想情况下, 每个处理器的运算时间相同, 我们称为负载均衡, load balance, 当每个处理器完成运算的时间有长有短时, 先完成运算的处理器必须等待未完成的处理器, 这样就相当于分配在每个处理器上的任务不同, 负载不同, 称为负载失衡, load imbalance. 显然负载失衡越小越好, 理想情况下是0, 这就是为什么gmx报告这个数字的原因, 你可以根据这个数字看看并行性能如何. 如果太大的话, 那说明你的并行效率有点差, 可能需要调整一些参数.
具体到gmx的并行, 负载失衡有很多方面的原因, 但其中最主要的是PP/PME分配不好引起的失衡. 这是因为gmx在计算时有两种模式, 一种是无论计算PP还是PME, 都使用全部的处理器, 另一种是只是用几个处理器单纯做PME计算, 剩下的处理器单纯做PP计算. 这两种模式哪种并行效率高, 需要测试才知道, 但一般对非均相体系, 如slab, 推荐使用后面的模式, 分配一些处理器单纯用于PME计算, 而其他的单纯用于PP计算. 可以调整设置使得PP/PME的负载失衡最小, 这样整体的负载失衡也会小一些, 并行效率高.
至于具体的mdrun的选项, 就是-dd, -npme, 可以参考程序的文档. 更具体的例子, 中文教程上有一篇, 但只是指导性的, 具体到你自己的计算, 还需要测试才知道.
此外, mdrun还有一个-dlb选项, 就是要不要开启自动负载均衡. 也就是mdrun的时候, 默认自动开始负载均衡. 具体就是根据运行前几百步的负载失衡数据, 如果数值很大, 就自动开启动态负载均衡dlb. -dlb yes则设定始终使用dlb, 无论数据大小. -dlb no就关闭了dlb
使用dlb的好处是并行效率可能高些, 但得到的轨迹没有二进制级别的可重复性, 也就是, 同一个输入文件, 使用dlb的话你运行两次, 得到的轨迹进行二进制级别的比较时不会完全一样. 理论上, 关闭dlb时同一输入文件运行两次得到的轨迹应该完全一样, 二进制级别的比较没有差异.
解释下, PP指particle-particle 粒子粒子相互作用的计算, 主要是非键相互作用, 计算量与粒子数平方成N^2正比, PME是用Ewald方法计算静电相互作用, 计算量和粒子数的对数NlnN(不是很确定)成正比, 前者的计算量大于后者, 所有设置时gmx的要求是Npme最大只能是总处理器数目的1/2. 具体多少, 可根据测试而定.
从实际使用来看,一般如果体系是均匀的,用dlb auto没什么问题,他分割盒子的时候每块的原子数差不多,计算量也差不多。但是如果体系里有大块的真空,就需要用dd手动指定盒子的分割方式了,尽量然分割的每块原子数差不多。



或许,我们是应该放弃一些固有的东西,并且应该承担起拯救自然的使命和责任了。因为,悲观和绝望无济于事——“我们实现明天理想的唯一障碍,就是对今天的疑虑。”
这里推送最新发布的招聘信息. 你可以看看有没有适合自己的位置, 或从中了解相关领域目前的研究项目.

本周刊记录我每周所读所思, 并自觉值得与大家分享的内容.
本周刊同步更新在我的网络日志 哲·科·文 和微信公众号 分子模拟之道.
如果你觉得我的分享对你有益, 不妨将它推荐给你认识的人.
如果你也认同分享的理念, 欢迎投稿或推荐自己的内容. 请关注微信公众号后台留言, 或加入QQ群联系.