在以前的一篇文章中, 我简单说过基于GROMACS的分段模拟方法. 这种方法非常通用, 几乎能完成任意的功能, 且无须修改源代码, 但是运行效率比较差, 因为每次运行mdrun都要重新生成tpr文件. 对大分子来说, 使用grompp生成tpr还是很耗时的, 可能会成为运行的瓶颈部分.
最近重新思考了一下这个问题, 想到, 在拓扑和模拟参数不变, 只有坐标或速度改变的情况下, 我们没有必要重新运行grompp生成tpr, 可以直接修改tpr中的坐标或速度, 然后使用修改过的tpr运行模拟. 这应该是不修改源代码情况下能做到的最快方法了. 与此类似的另一种方法是直接修改cpt文件, 可以达到同样的目的, 但适用情况有点不同.
tpr和edr文件都是二进制文件, 格式比较复杂, 完全弄明白且自如地修改它们并不容易, 但如果只是修改其中的一部分, 就要简单些.
对tpr的最简单修改应该是修改原子坐标了. 在体系拓扑不变的情况下, 利用外部程序修改体系中部分原子的坐标, 然后使用mdrun直接运行修改后的tpr文件, 就可以将mdrun当做一个引擎, 达到自己的目的.
这种方法的一个简单应用就是用GROMACS来做MC或对接, 刚性或柔性的都可以. 如果只是简单地计算下单点的能量, 就是刚性的; 如果进行能量最小化, 那就是柔性的. 更复杂的能量, 或许可以使用先进行能量最小化再模拟一段时间的平均能量, 甚至可以使用基于MM-PBSA的自由能. 这些都是可行的, 理论上也没有什么困难, 虽然效率可能不佳.
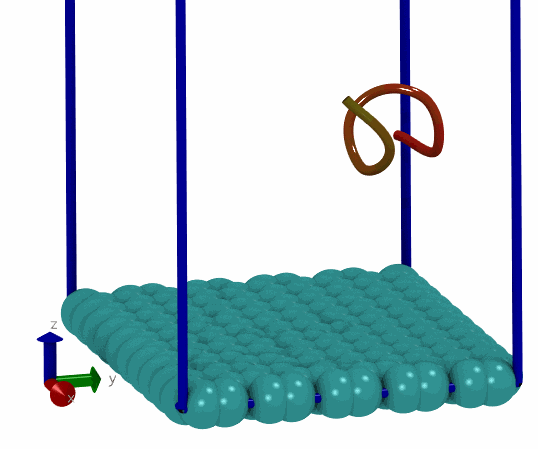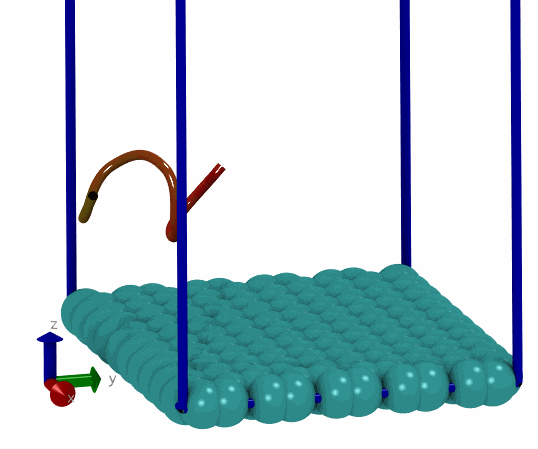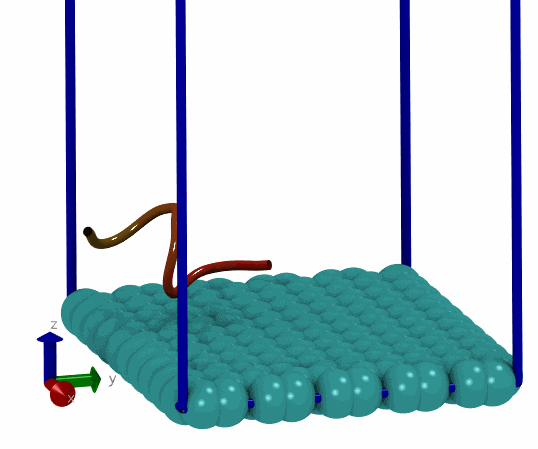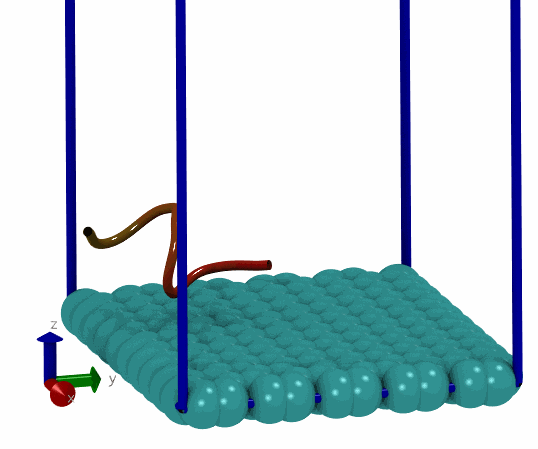
基于上面的想法, 我就尝试写了一个简单的MC程序, 用来搜索能量最低的构型. 拿一个小肽和石墨烯的体系做测试, 发现确实可以找到能量很低的构型. 这说明拿mdrun作为模拟引擎来实现自己的MC程序确实可行, 而且也比较简单, 只要关注MC移动部分就可以了, 能量计算, 构型优化都可以交由mdrun处理. 当然, 缺点在于效率可能不够好. 但对于普通体系来说, 效率可能也就够了, 没有必要弄得最高.