toc {:toc}
原作: Chris Lim and David A. Case, 原始文档
(译者注: 离子液体指全部由离子组成的液体, 常见的盐需要在高温下才呈现液态, 如KCl, KOH等, 而在室温或者室温附近呈液态的由离子构成的物质称为室温离子液体, 如本教程中提到的[bmim][BF4]室温离子液体.)
本教程的目的是指导使用Amber, AmberTools以及VMD的用户模拟室温离子液体并计算径向分布函数(RDF).
室温离子液体(RTILs), 正如它的名字所言, 是一种在室温下以液态存在的盐. RTILs引起各个研究领域的兴趣是因为它们具有独特的物理学和热力学性质: 如可忽略的蒸汽压, 高热稳定性, 高电化学稳定性以及不易燃等特点. 此外, 离子液体具有高电导和宽电化学窗口, 这意味着它们既不容易被氧化也不容易被还原, 因此它们是电池中高效和绿色电解质的优良候选物. 由于RTILs具有各种潜在应用价值, 关于RTILs的模拟工作也广泛的普及, 并设计和测试了各种阳离子和阴离子的组合, 以期拓展我们对这个领域的认识.
在本教程中, 我们将生成1-丁基-3-甲基咪唑四氟硼酸盐([bmim][BF4])和乙腈(CH3CN)来重现参考文献[1,2]中的数据. (译者注: 原文在这里添加参考文献的名称和期刊号, 我们直接放在参考文献部分, 并给出原文文献.)
提示: 该教程中用的是普适Amber力场(GAFF), 而不是文献中的优化过的力场. 模拟结果的某些部分与上述论文部分结果吻合的不是很好, 但这也给我们一个很好的启示: GAFF可以帮助你开始模拟, 并能够提供合理准确的结果, 从这点出发也许已经足够. 但如果你需要更为精准的结果, GAFF可以作为力场改进的基石, 这涉及特定的体系, 在该教程中不再详细描述.
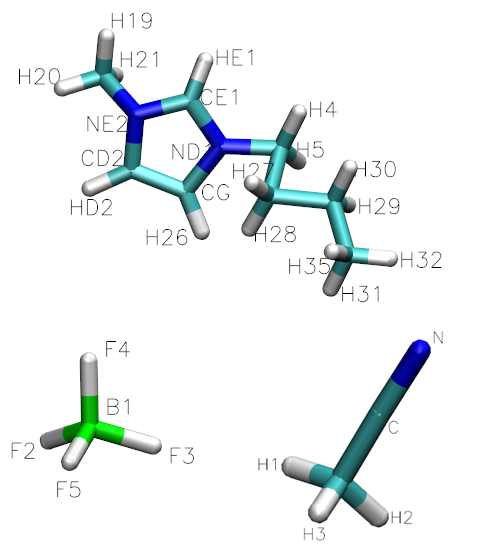
图1: 离子液体组分分子结构(上图为1-丁基-3-甲基咪唑, 即[bmim], 左下角图为四氟硼酸, 即[BF4], 右下角为乙腈, 即CH3CN)
xleap绘制分子我们用xleap绘制分子并用它产生pdb文件. 首先在命令窗口输入xleap, 弹出如下窗口(图2):
图2: xleap窗口
接下来, 我们将创建我们第一个组分, 在xleap命令窗中键入: edit BF4, 弹出如下窗口(图3):
图3: 弹出的分子绘制界面
这里我们将创建一个BF4分子, 所需的图形窗口弹出如图3所示. 绘制分子非常直观: 在Manipulation边框中勾选绘制(Draw)项, 选择合适的元素, 鼠标左键绘制一个原子, 然后拖动绘制一个键. 这里对于BF4, 我们从Elements栏目中从other elements下拉菜单中选择Boron元素, 然后左键绘制一个硼原子(图4所示).
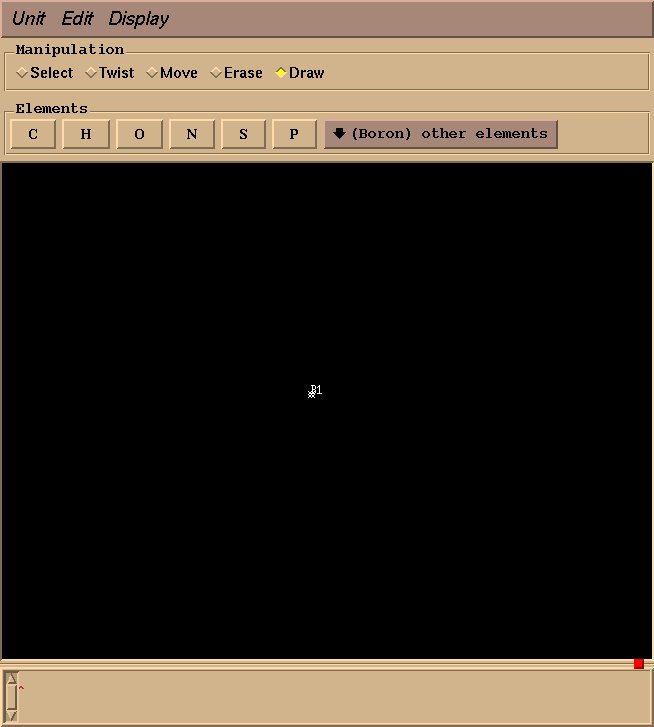
图4: 绘制了一个硼原子
紧接着, 在其他元素选项中(other elements)选择氟原子(Fluorine), 并且从硼原子出拖动鼠标到你所需放置氟原子的位置.
图5
用同样的方式拖动其他三个氟原子, 大概形成正四面体的结构即可. 这一步不需要完美的几何结构, 粗略估计已经足够(译者注: 后面能量最小化会正确优化结构).
提示: 摁住ctrl键并且左键移动鼠标旋转可以观察分子; 摁住ctrl并右击鼠标后, 向上移动放大分子, 向下移动则缩小分子. 如果单击选择好了一个原子, 摁住shift后单击绘图面板中的任意其他位置则取消选择(译者注: 释放shift键后才可以看到取消选择的状态).
图6
通过拖动鼠标在分子上方选择所有的原子(或通过单击单独的原子选择), 在Edit菜单下选择Relax selection选项后, xleap将为我们优化BF4分子的几何结构.
提示: 如果键盘上的NUM/CAPS(译者注: 数码锁定/大写锁定)锁打开的话菜单栏可能无法正常工作, 请注意关闭.
图7
关闭退出该窗口. 提示: 不要点击窗口右上角的X按钮, 因为这样会直接完全关闭xleap窗口, 而你的文件没有被保存, 所以要在Unit菜单栏下Close选项保存退出.
为生成我们所需的BF4分子, 在命令窗口中输入如下指令:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">savepdb</span> BF4 bf4.pdb </pre></div>重复步骤1和2创建剩下的两种分子.
(提示: 在本教程中, 我们用acn指代乙腈, bmi指代bmim, bf4指代BF4)
我们用leap产生包含电荷信息的mol2文件. 下面命令将为我们从pdb文件生成mol2文件, 在leap命令窗口中键入:
(-i表示输入文件, -fi是输入文件格式, -o为输出文件, -fo为输出文件格式, -c为所用电荷方法, -nc表示分子的净电荷. 不要忘记分别用-nc 1或者-nc -1来指定阳离子和阴离子的电荷.)
Antechamber不认识硼原子, 所以我们需要自己手动输入其参数. 我们用到的参考文献中给出了所需每个原子的的电荷, 所以我们将用到这些数值.
2.2.1 不带电荷模式运行antechamber
输入命令:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">antechamber</span> –i bf4.pdb –fi pdb –o bf4.mol2 –fo mol2 </pre></div>2.2.2 编辑mol2文件
采用你所喜欢的文本编辑器(如vi或gedit), 打开bf4.mol2文件, 我们需要修改以下几点:
第一, 如果第三行的第二个数字值为0, 把它修改成4. 这意味着我们的分子包含四个键(四个氟原子键连到硼原子).
第二, 定位到ATOM部分的最后一列, 这些数值代表了每个原子的电荷, 我们看到它们现在全部是0, 因为我们没有去指定它们. 随后用参考给出的这些值修改这列数值. (硼原子: 1.1504, 氟原子: -0.5376)
最后, 我们需要手动添加键信息. 键信息由指定的键id号, 成键的两个原子编号(根据mol2文件中ATOM原子编号而来), 以及键类型(1为单重建, 2为双重键等等)所组成.
具体来说, 我们将在BOND类别下首先添加这样一条的信息:
1 1 2 1
这条记录的意思是说第一条记录(第一个数值)是对B1原子(第二个数值)和F2原子(第三个数值)会形成单重建(第四个数值).
类似的, 添加另外三条记录:
1 1 3 1
1 1 4 1
1 1 5 1
提示: 键的id号全部是1, 因为键id号在这里是没有含义的.
xLEaP中输入:用antechamber生成bmim和BF4部分的mol2文件, 并用适当的名称替换输入名称和输出名称. 对每个分子重复步骤1和2. 这里你可以得到我们生成的mol2文件: acn.mol2, bmi.mol2和bf4.mol2, 并与你生成的文件进行比较.
Parmchk创建力场修正文件(frcmod)并补充缺少的参数. 以下命令可为先前创建的mol2文件生成一个frcmod文件:
对剩余的两个mol2文件重复此步骤. 可以在这里得到我们生成的frcmod文件: frcmod.acn, frcmod.bmi和frcmod.bf4. 你可以将它们与你自己所生成的frcmod文件相比较. 请注意, frcmod.acn文件基本上是空的, 因为该分子所需的所有参数都在gaff.dat文件中. 如果你愿意, 也可以不用这些frcmod文件.
Packmol可以在给定范围内排布组装分子, 为分子动力学模拟提供起始原子坐标. Packmol程序考虑了短程排斥相互作用从而保证了在模拟中体系不会奔溃
1. 在http://www.ime.unicamp.br/~martinez/packmol/下载Packmol并按照安装说明进行安装.
2. 创建一个名为input.inp的输入文件, 例如:
3. 运行Packmol
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">packmol</span> < input.inp </pre></div>4. 在可视化分子动力学软件(VMD)中查看由packmol生成的pdb文件:
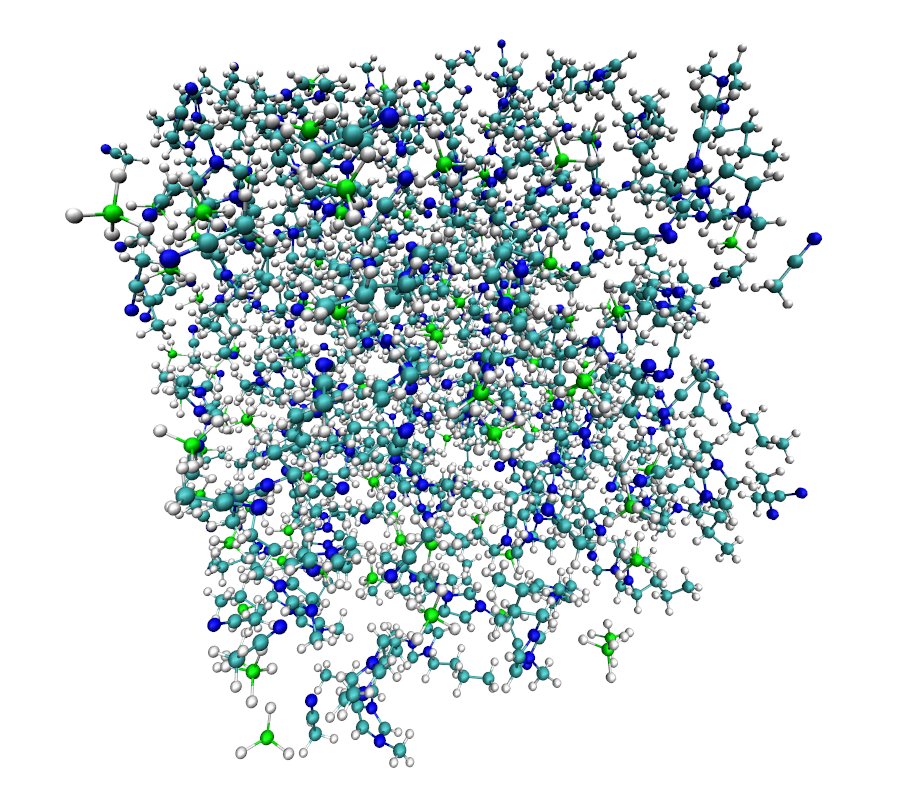
图8: packmol生成的结构
prmtop文件用tLEaP为刚才packmol中产生的pdb文件生成参数拓扑文件(prmtop)和坐标文件(inpcrd).
1. 创建一个输入文件(名为commands.in):
2. 用输入文件(commans.in)运行tLEaP:
创建一个名为runmin.sh的脚本. 该脚本将创建输入文件并运行sander. imin = 1告诉sander进行最小化, ntpr = 100表示每100步保存一次重启文件, ntwx = 100表示每100步输出一次轨迹, maxcyc = 10000表示最小化循环10000次, ntb = 1指定周期性边界条件.
注意: 如果要第二次运行模拟, 请确保修改sander的参数. 例如, min1完成后, 第二次输入坐标文件应为min1.x, 而输出文件则应以min2开头.
用以下命令运行这个程序:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">./runmin.sh</span> </pre></div>你应当查看Amber手册以了解更多有关这些参数的信息.
创建名为runmd.sh的脚本文件
再次提醒, 你应该看看Amber手册, 以了解这个输入文件到底在做什么.
注意: MD模拟产生的重启文件中包含速度信息, 而最小化的启动文件没有包含速度信息. 为了标记这些差异性, 使用ntx = 5和irest = 1标记重启文件, 以便在后面的模拟中区分这些重启文件.
另请注意: 当你运行下一个模拟时, 请使用上一个输出中的重启文件作为新的输入坐标文件.
ptraj成像我们用packmol创建的盒子显示出一个基本问题: 大部分分子暴露于真空, 这会使模拟结果产生偏差. 在周期性边界下, 我们的盒子在所有三个维度都会周期性"复制", 以使我们的系统代表真实的散装液体环境. 因此, 当一个分子从一侧离开盒子时, 它会从另一侧进入. Ptraj的镜像工具重新调整了离开盒子的分子, 为我们提供了中心盒子中的正确分子构象.
创建一个输入文件ptraj.in:
要运行ptraj, 请运行以下命令:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">ptraj</span> prmtop ptraj.in </pre></div>Ptmtop是我们盒子的拓扑文件.

图9: MD结果:(左)在ptraj处理之前; (右)ptraj处理之后
径向分布函数(RDFs)
1. 首先分析密度, 看平均密度是否靠近目标值.
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">awk</span> '/Density/ { print $3 }; $1=="A" && $2=="V" {exit 0}' md1.out > density.dat </pre></div>这个awk脚本计算输入文件(md1.out), 并输出每行出现Density一词的第三列. 密度输出到density.dat文件中. 当awk到达输出文件中的"平均"部分时, 脚本退出.
2. 使用xmgrace绘制密度数据图:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">xmgrace</span> density.dat </pre></div>3. 要计算平均密度, 请检查xmgrace中的设置, 或运行以下脚本:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">awk</span> '{ sum = sum + $1 }; END { print "average = ", sum/NR }' density.dat </pre></div>这个awk脚本利用density.dat中的数据, 并将每行的第一列添加到sum中, 然后输出除以NR(awk中定义行数的变量)的总和得到平均值.
4. 检查Energy Total以查看系统是否处于平衡状态.
如果要使用xmgrace绘制Energy Total数据图, 请使用与上述绘制密度数据图类似的命令.
以下是[bmim] [BF4]和乙腈的离子液体混合物的MD模拟的Energy Total图示例:
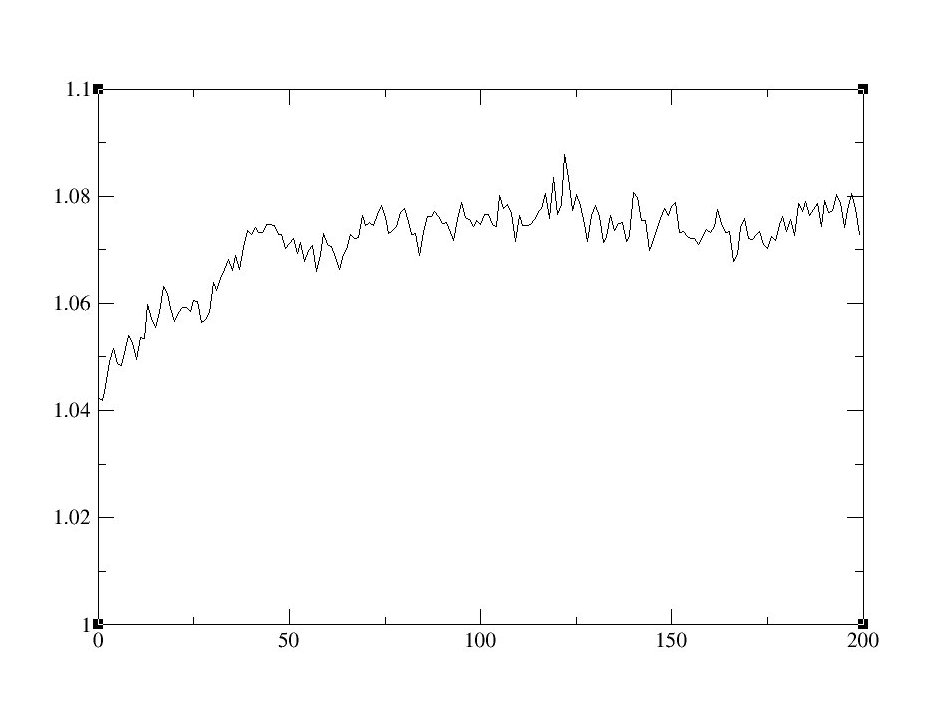
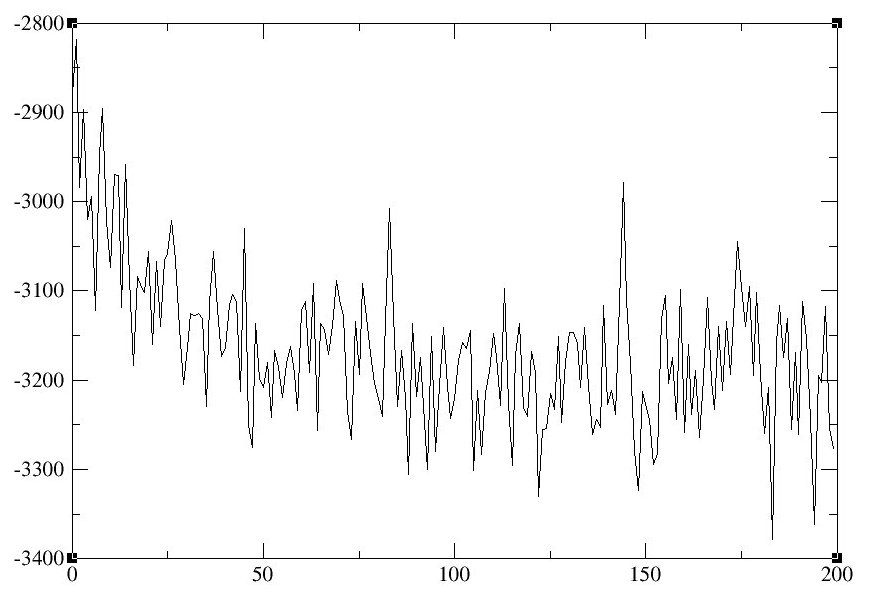
图10: 密度(左)和总能量(右)随时间的变化曲线.
5. 一旦密度达到目标值且总能量处于平衡状态, 便可以计算径向分布函数.
这里得到的平均密度为1.0774cc/mol, 参考文献中的密度1.087cc/mol.
为了计算径向分布函数, 我们将使用ptraj, 这是一个用于分析输入坐标文件中的三维坐标的程序. 我们将计算乙腈[CH3CN]的N1原子的径向分布函数.
1) 创建一个输入文件(ptraj.in):
2) 运行
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">ptraj</span> prmtop ptraj.in </pre></div>CH3CN_N1是输出文件名称的标题, .1(指的是0.1, 译者注)是计算间隔的大小, 15.0是直方图的最大值, 以及:ACN@N是用于选择我们想要用于分析的原子的筛选命令(即acn分子中的N原子).
运行ptraj.in文件后生成的输出文件将为: CH3CN_N1_carnal.xmgr, CH3CN_N1_standard.xmgr CH3CN_N1_volume.xmgr
使用Xmgrace打开文件以查看RDF的结果图. 例如:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">xmgrace</span> CH3CN_N1_volume.xmgr </pre></div>上述命令将打开xmgrace中的CH3CN_N1_volume.xmgr文件. x轴是以埃为单位的距离, 并且g(r)表示在一个原子(乙腈中的一个氮)的给定距离(r)处找到原子(在这种情况下为另一个乙腈中的氮)的概率. 第一个峰(约四埃)代表N原子的第一个聚集层, 第二个峰(少于六埃)代表N原子的第二个聚集层.
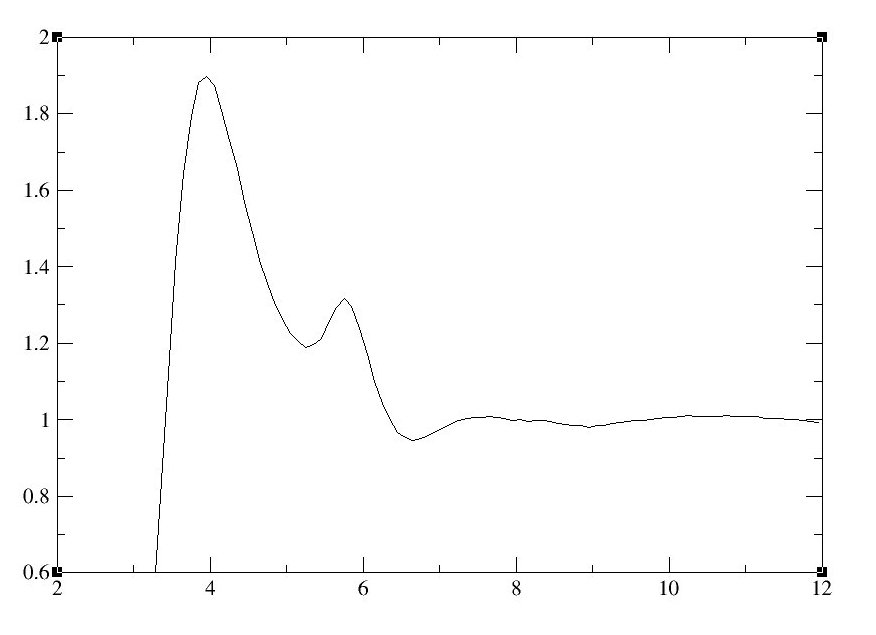
图11: 径向分布函数
根据爱因斯坦关系, 自扩散系数D可以用下面的公式计算:
$$Di = {1 \over 6} \lim{t \to \infty} {d\over dt} \lt [\vec r_i(t) -\vec r_i(0)]^2 \gt$$
其中 $D_i$ 表示每个组分的自扩散系数, $\lt \gt$ 符号中表示为均方根位移, 即MSD, 而 $\vec r_i(t)$ 表示其质心的的坐标向量, $\vec r_i(0)$ 为初始时刻的质心坐标向量. (译者注: 教程中的公式应该是由于网页转换公式出现了问题, 这里按照参考文献1给出的公式重新改写)
为了计算自扩散系数D, 我们将
1) 使用ptraj和gnuplot绘制平均MSD随时间的图
2) 找出图的斜率
3) 将值代入方程得到D
Ptraj将计算MSD并且输出x(时间)和y(MSD)到gnuplot的文件中.
创建名为ptraj.in的文件并输入以下内容:
mask这里表示筛选分子所需的命令. 例如, 如果您想为乙腈计算D, 请将mask替换为:ACN. 要为所有残基计算D(这是本教程的内容), 请使用:*.
Timestep为dt * ntwx, 是输出轨迹的时间间隔(以皮秒为单位).
运行以下命令:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">ptraj</span> nameOfYourTopologyFile ptraj.in </pre></div>打开gnuplot(在shell中输入gnuplot). 你应该在提示信息后出现后的一行左侧看到>, 这意味着我们已经可以运行gnuplot命令了.
图12
输入:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">plot</span> 'diffusion_r.xmgr' with lines </pre></div>使用该图作为参考, 计算图的斜率并将该值代入到公式中. 斜率的单位是平方埃每皮秒.
注意: 虽然公式表明应该采用图形末尾附近的斜率, 但ptraj的扩散工具在图形的开头是最准确的. 这是显而易见的, 因为噪声似乎随着x的增加而增加.
另请注意: 模拟中恒温和恒压设置可能会破坏扩散系数. 为了获得更精确的测量结果, 请关闭温度和压强控制再模拟一次.
这里我们得斜率得为0.0635990平方埃每皮秒(或者是63.5990×10^-11^平方米每秒(m/s)). 为了得到扩散系数, 我们需要将这个数字除以6, 即我们得到的扩散系数为10.5998×10^-11^米平方每秒(m/s). 而参考文献的扩散常数为14.9×10^-11^米平方每秒(m/s), 这种差异的可能原因是我们用的是Amber普适力场, 而参考文献使用的是修正过的精细力场.
我们模拟室温离子液体时, 使用xleap"手动"绘制分子, 使用antechamber优化几何结构, 使用xleap/tleap生成拓扑和坐标文件, 并使用sander进行最小化和MD模拟. 另外, 我们还使用ptraj对我们收集的数据进行分析, 例如计算径向分布函数(RDF)和自扩散系数(self-diffusion coefficients), 并且我们使用xmgrace和gnuplot来帮助我们可视化数据. 希望在本教程结束时, 你会对AMBER组件提供的众多工具感到更加得心应手, 现在你可以继续进行更复杂的模拟(以及更复杂的分析!).