
脂类分子是磷脂双分子膜的重要组成部分, 在细胞信号和生理学过程中发挥着重要的作用. 如今Amber这类凝聚态分子动力学模拟软件已经可以用来模拟各种各样的生物分子, 包括脂类.
在本教程中, 我们将为读者展现建立磷脂双分子层的详细步骤, 以及如何使用Amber和Lipid14力场进行模拟. 我们假定读者此时已经在Unix系统下安装好了Amber14软件包, 并且对如何使用Amber有基本的了解.
本教程将为读者展示如何模拟磷脂双分子层以及带有膜结合蛋白的磷脂双分子层. 经验表明, 在处理蛋白系统前理解磷脂双分子层的动力学过程往往是很重要的. 接下来本教程会提供给读者关于使用Amber和Lipid14搭建膜结合蛋白系统的细节.
注意事项: 本教程中的内容仅作为指导作用, 此教程里的参数设置仅为了此教程选取, 对其他系统并不一定适用. 深刻理解每一步模拟将对构建其他系统有很大帮助.
charmmlipid2amber.py: 包含在AmberTools 14 update1内在使用Lipid14力场时, 请引用以下文章:
Dickson, C.J., Madej, B.D., Skjevik, A.A., Betz, R.M., Teigen, K., Gould, I.R., Walker, R.C., "Lipid14: The Amber Lipid Force Field", J. Chem. Theory Comput., 2014, 10(2), 865-879. DOI: 10.1021/ct4010307
Amber14包括Lipid14[1], 一个为无张力脂类模拟而设计的模型化的脂类力场. Lipid14包括Lipid11里的电荷模型[2], 以及GAFFlipid里对关键二面角和范德华力的再参数化[3]. Lipid14已经通过了广泛的测试, 在六种主要的脂质双分子层类型中得到验证. Lipid14参数化的策略是与其它对式加和的Amber力场的方法是一致和兼容的. 因此, 原则上Lipid14与Amber14里其他的生物分子力场完全兼容.
下表列出了目前支持的脂类"残基"类型, 一并列出的还有支持的脂类双分子层. 作为参考, 我们一并列出了之前版本中出现的脂类(GAFFlipid以及Lipid14). 另加的头部基团和尾部基团以后其他的双分子层成分如胆固醇将出现在Lipid14力场的后续更新中.
<table id='tab-0'><caption></caption> <tr> <th rowspan="1" colspan="3" style="text-align:center;">Lipid14脂残基</th> </tr> <tr> <th rowspan="1" colspan="1" style="text-align:center;">基团</th> <th rowspan="1" colspan="1" style="text-align:center;">说明</th> <th rowspan="1" colspan="1" style="text-align:center;">LIPID14残基名称</th> </tr> <tr> <td rowspan="5" colspan="1" style="text-align:center;">酰基链</td> <td rowspan="1" colspan="1" style="text-align:center;">Lauroyl (12:0)</td> <td rowspan="1" colspan="1" style="text-align:center;">LA</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Myristoyl (14:0)</td> <td rowspan="1" colspan="1" style="text-align:center;">MY</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Palmitoyl (16:0)</td> <td rowspan="1" colspan="1" style="text-align:center;">PA</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Stearoyl (18:0)</td> <td rowspan="1" colspan="1" style="text-align:center;">ST</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Oleoyl (18:1 n-9)</td> <td rowspan="1" colspan="1" style="text-align:center;">OL</td> </tr> <tr> <td rowspan="2" colspan="1" style="text-align:center;">头基</td> <td rowspan="1" colspan="1" style="text-align:center;">Phosphatidylcholine</td> <td rowspan="1" colspan="1" style="text-align:center;">PC</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Phosphatidylethanolamine</td> <td rowspan="1" colspan="1" style="text-align:center;">PE</td> </tr> <tr> <th rowspan="1" colspan="3" style="text-align:center;">Lipid11脂残基</th> </tr> <tr> <th rowspan="1" colspan="1" style="text-align:center;">基团</th> <th rowspan="1" colspan="1" style="text-align:center;">说明</th> <th rowspan="1" colspan="1" style="text-align:center;">LIPID11残基名称</th> </tr> <tr> <td rowspan="7" colspan="1" style="text-align:center;">酰基链</td> <td rowspan="1" colspan="1" style="text-align:center;">Palmitoyl (16:0)</td> <td rowspan="1" colspan="1" style="text-align:center;">PA</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Stearoyl (18:0)</td> <td rowspan="1" colspan="1" style="text-align:center;">ST</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Oleoyl (18:1 n-9)</td> <td rowspan="1" colspan="1" style="text-align:center;">OL</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Linoleoyl (18:2 n-6)</td> <td rowspan="1" colspan="1" style="text-align:center;">LEO</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Linolenoyl (18:3 n-3)</td> <td rowspan="1" colspan="1" style="text-align:center;">LEN</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Arachidonoyl (20:4 n-6)</td> <td rowspan="1" colspan="1" style="text-align:center;">AR</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Docosahexanoyl (22:6 n-3)</td> <td rowspan="1" colspan="1" style="text-align:center;">DHA</td> </tr> <tr> <td rowspan="7" colspan="1" style="text-align:center;">头基</td> <td rowspan="1" colspan="1" style="text-align:center;">Phosphatidylcholine</td> <td rowspan="1" colspan="1" style="text-align:center;">PC</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Phosphatidylethanolamine</td> <td rowspan="1" colspan="1" style="text-align:center;">PE</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Phosphatidylserine</td> <td rowspan="1" colspan="1" style="text-align:center;">PS</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Phosphatidic acid (PHO4 -)</td> <td rowspan="1" colspan="1" style="text-align:center;">PH-</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Phosphatidic acid (PO4 2-)</td> <td rowspan="1" colspan="1" style="text-align:center;">P2-</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">R-phosphatidylglycerol</td> <td rowspan="1" colspan="1" style="text-align:center;">PGR</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">S-phosphatidylglycerol</td> <td rowspan="1" colspan="1" style="text-align:center;">PGS</td> </tr> <tr> <td rowspan="2" colspan="1" style="text-align:center;">其他</td> <td rowspan="1" colspan="1" style="text-align:center;">Phosphatidylinositol</td> <td rowspan="1" colspan="1" style="text-align:center;">PI</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">Cholesterol</td> <td rowspan="1" colspan="1" style="text-align:center;">CHL</td> </tr> <tr> <th rowspan="1" colspan="3" style="text-align:center;">GAFFlipid 分子</th> </tr> <tr> <th rowspan="1" colspan="2" style="text-align:center;">名称</th> <th rowspan="1" colspan="1" style="text-align:center;">GAFFlipid分子名称</th> </tr> <tr> <td rowspan="1" colspan="2" style="text-align:center;">1,2-dilauroyl-sn-glycero-3-phosphocholine</td> <td rowspan="1" colspan="1" style="text-align:center;">DLPC</td> </tr> <tr> <td rowspan="1" colspan="2" style="text-align:center;">1,2-dimyristoyl-sn-glycero-3-phosphocholine</td> <td rowspan="1" colspan="1" style="text-align:center;">DMPC</td> </tr> <tr> <td rowspan="1" colspan="2" style="text-align:center;">1,2-dioleoyl-sn-glycero-3-phosphocholine</td> <td rowspan="1" colspan="1" style="text-align:center;">DOPC</td> </tr> <tr> <td rowspan="1" colspan="2" style="text-align:center;">1,2-dipalmitoyl-sn-glycero-3-phosphocholine</td> <td rowspan="1" colspan="1" style="text-align:center;">DPPC</td> </tr> <tr> <td rowspan="1" colspan="2" style="text-align:center;">1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine</td> <td rowspan="1" colspan="1" style="text-align:center;">POPC</td> </tr> <tr> <td rowspan="1" colspan="2" style="text-align:center;">1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine</td> <td rowspan="1" colspan="1" style="text-align:center;">POPE</td> </tr> </table>在开始磷脂双分子层模拟之前, 构建一个可供Amber的LEaP程序处理的PDB格式的初始构型是很重要的. 构建结构有以下几种方法:
在此教程中, 我们使用CHARMM-GUI网站来搭建我们的磷脂双分子膜结构, 然后将此PDB文件转换成可供Amber程序LEaP读取的文件格式. 为了简便起见, 我们将用此网站搭建一个初始的磷脂双分子膜结构. 本教程我们用128个脂类基团以及Lipid14力场来搭建一个简单的DOPC磷脂双分子结构, 来进行后续的MD模拟.
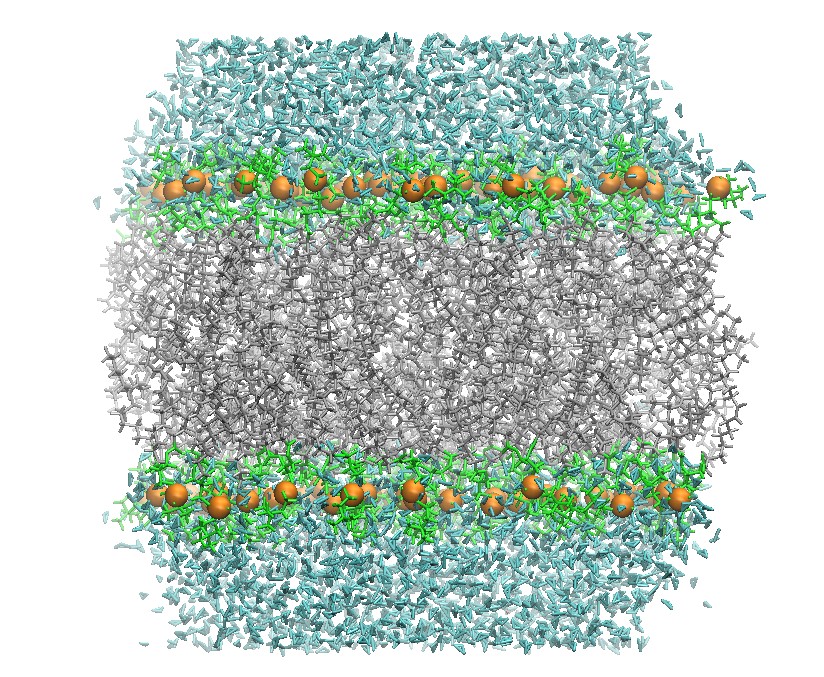
128个DOPC磷脂双分子层的截面图如下
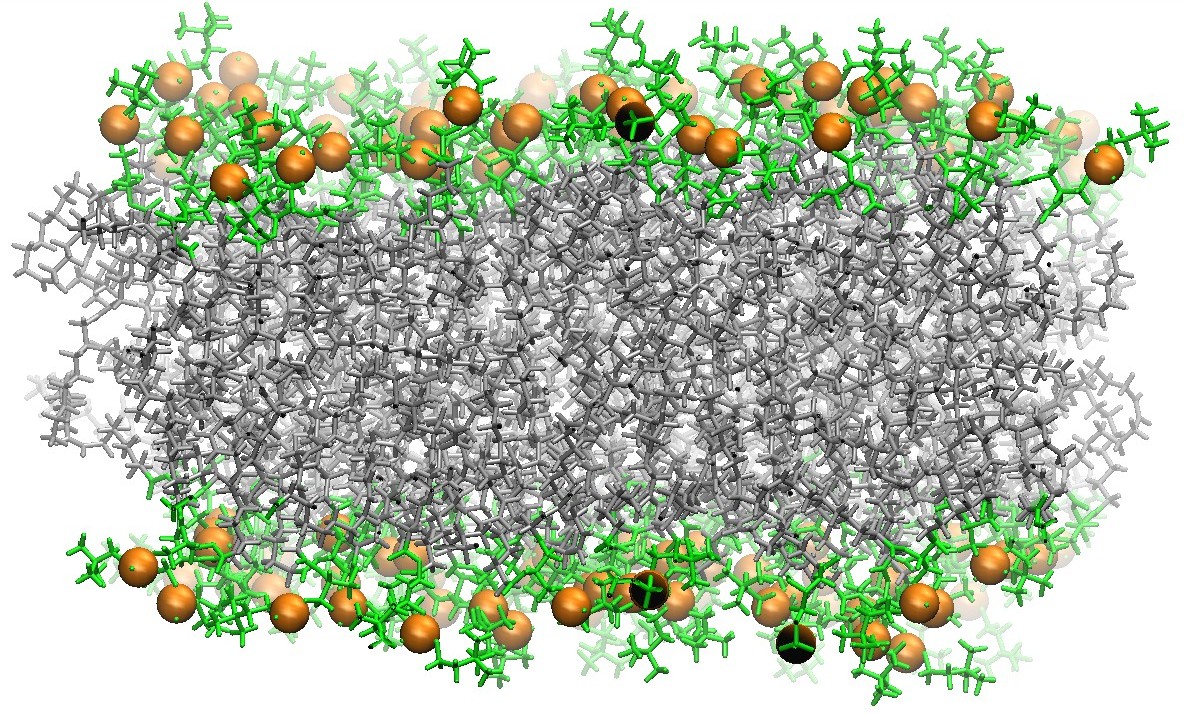
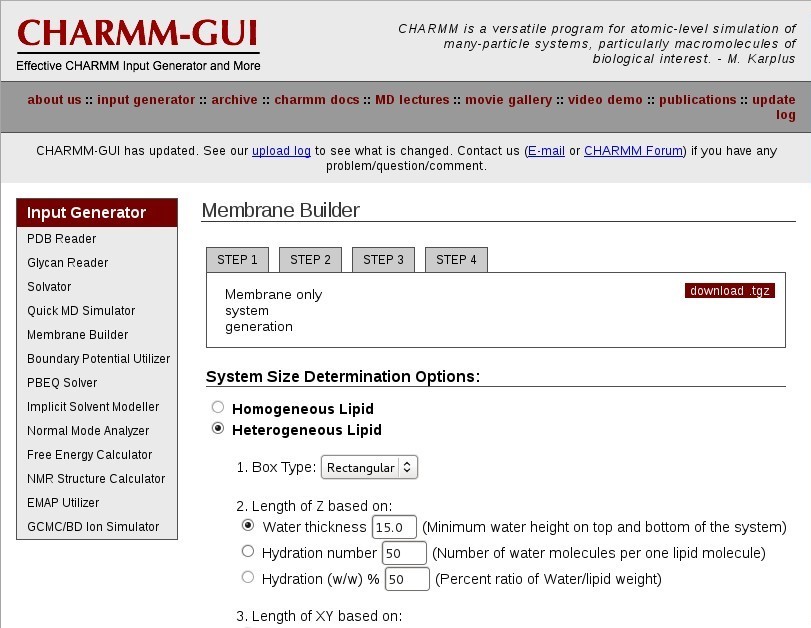
选择Membrane only system(仅包含膜的系统), 点击Next step.
选择Heterogeneous Lipid(非均匀脂分子)系统.
Box type: 选择Rectangular这样做会生成一个立方型, 并在X, Y, Z轴上具有周期性边界条件的系统.
这一步很重要因为你需要一个合适的水溶膜环境. 在此我们建议你参考一下关于针对不同脂类的水化数目的文献以得到一个合适的个数.
对我们的DOPC来说, 选择中间一项Hydration number, 设定每个脂分子对应37个水分子. 此数值稍大于实验结果32.8[4].
Number of lipid components这一步是为了定义XY方向的长度. 这样做的话程序会根据每个脂分子的表面积来构建系统.
Lipid Type列表第三项PC(phosphatidylcholine) Lipids下的DOPC类型后的# of Lipid on Upper leaflet和# of Lipid on Lower leaflet两个文本框里输入脂分子数目64.# of Lipid on Upper leaflet的意思是上层的脂类分子数目. 对于我们的DOPC系统来说两层都是64.
在CHARMM-GUI table中你可以找到CHARMM-GUI支持的脂类列表.
<table id='tab-1'><caption>CHARMM-GUI目前支持的脂分子与Lipid14残基</caption> <tr> <th rowspan="1" colspan="1" style="text-align:center;">CHARMM-GUI脂分子</th> <th rowspan="1" colspan="1" style="text-align:center;">Lipid14残基1</th> <th rowspan="1" colspan="1" style="text-align:center;">Lipid14残基2</th> <th rowspan="1" colspan="1" style="text-align:center;">Lipid14残基3</th> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">DLPC</td> <td rowspan="1" colspan="1" style="text-align:center;">LA</td> <td rowspan="1" colspan="1" style="text-align:center;">PC</td> <td rowspan="1" colspan="1" style="text-align:center;">LA</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">DMPC</td> <td rowspan="1" colspan="1" style="text-align:center;">MY</td> <td rowspan="1" colspan="1" style="text-align:center;">PC</td> <td rowspan="1" colspan="1" style="text-align:center;">MY</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">DPPC</td> <td rowspan="1" colspan="1" style="text-align:center;">PA</td> <td rowspan="1" colspan="1" style="text-align:center;">PC</td> <td rowspan="1" colspan="1" style="text-align:center;">PA</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">DOPC</td> <td rowspan="1" colspan="1" style="text-align:center;">OL</td> <td rowspan="1" colspan="1" style="text-align:center;">PC</td> <td rowspan="1" colspan="1" style="text-align:center;">OL</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">POPC<sup>*</sup></td> <td rowspan="1" colspan="1" style="text-align:center;">PA</td> <td rowspan="1" colspan="1" style="text-align:center;">PC</td> <td rowspan="1" colspan="1" style="text-align:center;">OL</td> </tr> <tr> <td rowspan="1" colspan="4" style="text-align:center;"></td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">DLPE</td> <td rowspan="1" colspan="1" style="text-align:center;">LA</td> <td rowspan="1" colspan="1" style="text-align:center;">PE</td> <td rowspan="1" colspan="1" style="text-align:center;">LA</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">DMPE</td> <td rowspan="1" colspan="1" style="text-align:center;">MY</td> <td rowspan="1" colspan="1" style="text-align:center;">PE</td> <td rowspan="1" colspan="1" style="text-align:center;">MY</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">DPPE</td> <td rowspan="1" colspan="1" style="text-align:center;">PA</td> <td rowspan="1" colspan="1" style="text-align:center;">PE</td> <td rowspan="1" colspan="1" style="text-align:center;">PA</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">DOPE</td> <td rowspan="1" colspan="1" style="text-align:center;">OL</td> <td rowspan="1" colspan="1" style="text-align:center;">PE</td> <td rowspan="1" colspan="1" style="text-align:center;">OL</td> </tr> <tr> <td rowspan="1" colspan="1" style="text-align:center;">POPE<sup>*</sup></td> <td rowspan="1" colspan="1" style="text-align:center;">PA</td> <td rowspan="1" colspan="1" style="text-align:center;">PE</td> <td rowspan="1" colspan="1" style="text-align:center;">OL</td> </tr> <tfoot><tr><td colspan="4" style="text-align:left"> <sup>*</sup> 可以使用VMD构建并利用<code>charmmlipid2amber.py</code>进行处理<br> </td></tr></tfoot> </table>show the system info, 确认系统成分是否正确, 然后继续下一步.取决于你所需要的脂分子类型以及溶液环境, 往往我们会有离子浓度方面的需求. 对于脂类来说, 头部基团的固有电荷会和水溶液环境相互作用, 所以加入离子是非常重要的. 对于特定体系, 有时候对离子浓度有特殊的需求, 比如离子通道蛋白体系.
对于DOPC系统, 将离子浓度参数设为0.15 M KCL, Ion Placing Method选择Monte-Carlo方式来加入离子.
后两步将脂类双分子层系统生成单个结构文件
点击Continue to the next step
点击Continue to the next step
到了这一步, 脂分子, 水分子和离子已经被合成了一个PDB结构文件.
下载生成的.tgz文件, 保存到你的电脑里
在下载的charmm-gui.tgz压缩文件中, 我们只需要step_5assembly.pdb文件. 这个文件应当包含磷脂双分子层, 水分子, 以及离子. 确认没有丢失任何成分是非常重要的. 将step5_assembly.pdb保存到工作目录下.
CHARMM-GUI给出的结构如下
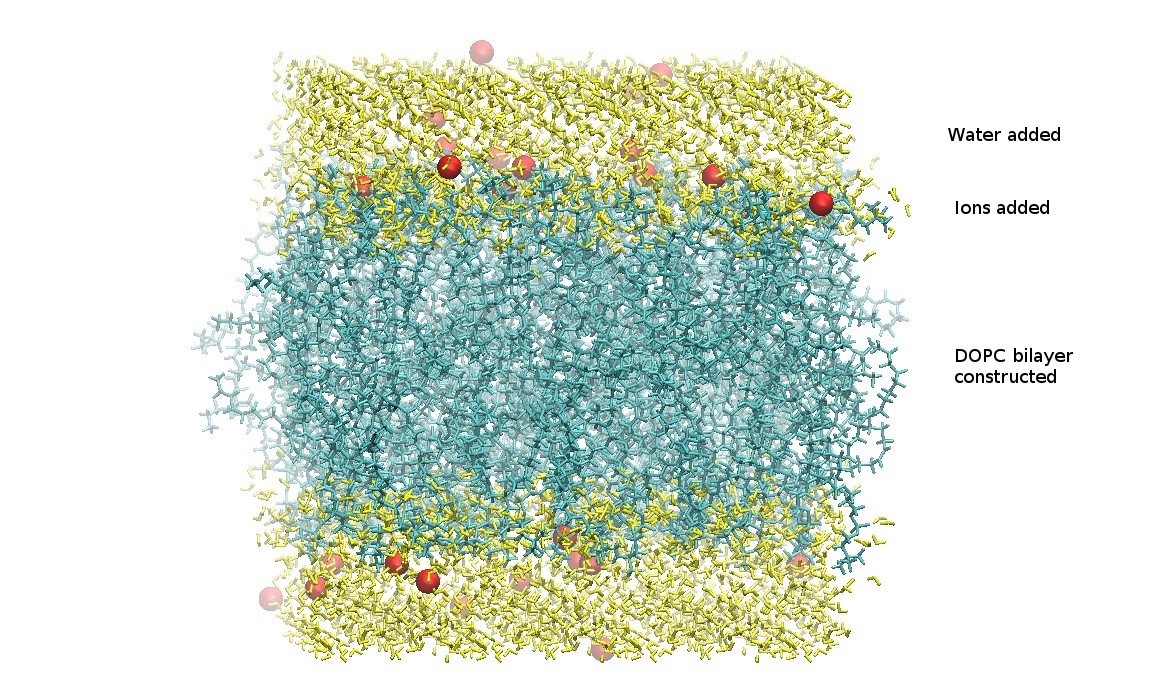
现在你有了磷脂双分子层的原始构型, 但是, 该PDB结构文件并不是所需要的标准格式.
简单的讲, Lipid14格式如下: 脂类被分成三个residue(残基): 两个尾部残基和一个头部残基. 在Lipid14中每个残基都有自己特定的残基名称, 原子名称和原子类型. 在一个Lipid14 PDB文件中, 一个磷脂分子由上述三种残基依次列出, 遵循如下顺序: sn-1尾基, 头基, sn-2尾基. 每个包含三种残基的脂分子必须以TER标签结尾.
Lipid14 PDB文件格式
# Phospholipid 1
Acyl chain 1 residue
Head residue
Acly chain 2 residue
TER card
# Phospholipid 2
Acyl chain 1 residue
Head residue
Acly chain 2 residue
TER card
...
LEaP要求所有CHARMM-GUI PDB格式的文件必须转换成Lipid14格式. 这可以通过下面的charmmlipid2amber.py来完成.
charmmlipid2amber.py文件是AmberTools 14升级包里的一个强制残基原子重命名及重排序脚本. 在这里我们用它来将CHARMM-GUI PDB格式的结构转化成可供Lipid14读取的PDB格式. 如果你没有Ambertools 14 update1, 你可以从上面的链接里下载这个文件. 下载后将它保存到Amber的目录下解压.
警告: 在ambertools14 update1中, charmmlipid2amber.py已经取代了charmmlipid2amber.x脚本.
现在, 使用charmmlipid2amber.py脚本将我们的CHARMM-GUI格式PDB文件转换为Lipid14格式PDB文件.
用法:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">charmmlipid2amber.py</span> <span style="color:#666">-i</span> input_structure.pdb [-c substitution_definitions.csv] <span style="color:#666">-o</span> output_structure.pdb </pre></div>其中input_structure.pdb是CHARMM-GUI格式PDB文件, output_structure.pdb是可供LEaP读取的Lipid14格式的PDB文件.
对我们的系统来说:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">charmmlipid2amber.py</span> <span style="color:#666">-i</span> step5_assembly.pdb <span style="color:#666">-c</span> $AMBERHOME/AmberTools/src/etc/charmmlipid2amber/charmmlipid2amber.csv <span style="color:#666">-o</span> DOPC_128.pdb </pre></div>脚本执行完之后, 你会得到一个名为DOPC_128.pdb的文件, 接下来我们可以用LEaP载入这个文件了.
由于CHARMM-GUI构建脂类分子层的方法所限, 在XY方向上会有脂类分子超出水溶液层的边界. 我们通过水分子的坐标来测量预估盒子尺寸的话得到更好的结果. 我们可以用PDB结构可视化程序如VMD来手动测量盒子尺寸.
这里我们提供一个简单的bash/vmd脚本vmd_box_dims.sh来测量周期性盒子的尺寸.
使用方法:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">vmd_box_dims.sh</span> <span style="color:#666">-i</span> input_structure.pdb <span style="color:#666">-s</span> vmd_selection </pre></div>现在, 利用水分子位置来计算盒子尺寸
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">vmd_box_dims.sh</span> <span style="color:#666">-i</span> DOPC_128.pdb <span style="color:#666">-s</span> water <span style="color:#A2F">81.35599899291992</span> 80.00199890136719 70.54999923706055 </pre></div>在这一节里, 我们将把DOPC_128.pdb载入LEaP里来定义脂类残基的拓扑结构和参数. 使用以下方法来准备Amber的拓扑参数文件和初始的坐标文件.
启动xLEaP
在LEaP命令行中, 指定你想使用的力场. 此例中我们使用Lipid14
载入力场:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">source</span> leaprc.Lipid14 <span style="color:#A2F">source</span> leaprc.ff12SB <span style="color:#A2F">loadamberparams</span> frcmod.ionsjc_tip3p </pre></div>LEaP载入DOPC_128.pdb中包含的结构, 将脂类分成三部分, 并指定原子类型.
合成脂类结构文件
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">DOPC</span> = loadpdb DOPC_128.pdb </pre></div>注意: 如果出现对于脂类被分成不同的残基的警告, 不用担心, 这是LEaP的正常行为. 残基的序列号可能会和PDB文件中的不同.
现在我们使用刚刚测出的数据作为盒子尺寸
为系统添加周期性盒子
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">set</span> DOPC box { 81.35599899291992 80.00199890136719 70.54999923706055 } </pre></div>保存用于分子模拟的Amber prmtop拓扑及参数文件和inpcrd原始坐标文件.
注意: 保存参数文件及拓扑文件时, 仔细留意amber的错误提示. 检查有没有遗漏的参数.
到了这一步, 我们已经有了模拟所需要的文件: DOPC_128.prmtop, DOPC_128.inpcrd
退出LEaP, 确认我们已经创建好.prmtop以及.inpcrd文件.
128个DOPC脂双分子层的初始结构
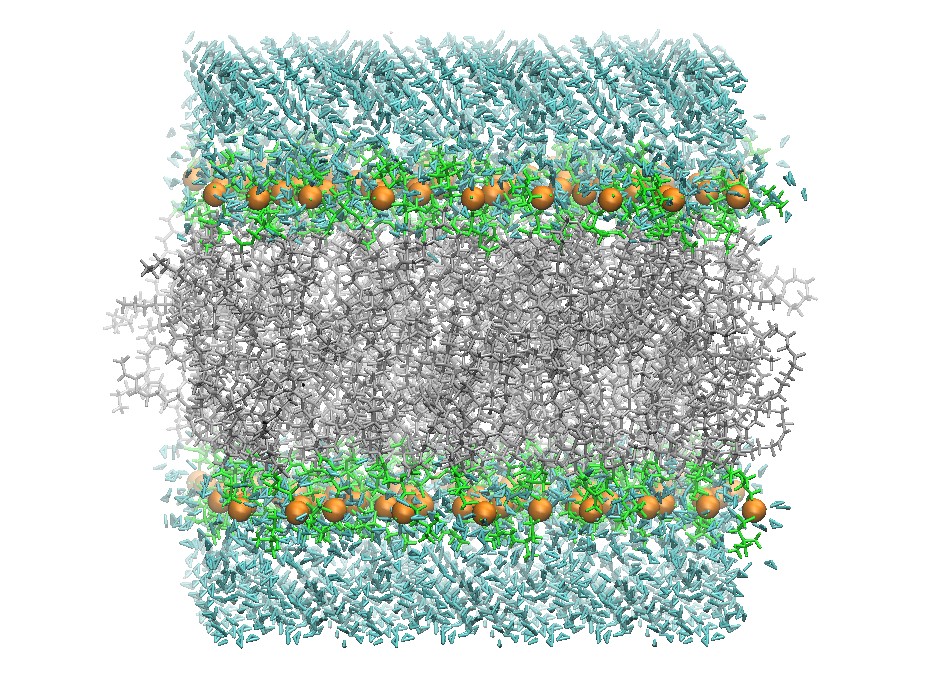
警告: 脂类双分子层的分子动力学模拟并不简单. 在开始之前, 我们建议你先参考一下相关的文献资料. 以下我们使用的方法和在Lipid14发表文献中使用的相似.
对于脂类双分子层的模拟, 我们采取以下步骤:
第一步的能量最小化非常重要, 因为初始的结构需要经历能量最小化来达消除异常构型来达到平衡. 以下能量最小化方法在Amber模拟中非常普遍.
注意: 为了更好的解释每一步, 以下的文件包含注释
<table class="highlighttable"><th colspan="2" style="text-align:left">01_Min.in</th><tr><td><div class="linenodiv" style="background-color: #f0f0f0; padding-right: 10px"><pre style="line-height: 125%"> 1 2 3 4 5 6 7 8 9 10 11 12 13</pre></div></td><td class="code"><div class="highlight"><pre style="line-height:125%"><span></span>Lipid minimization <span style="color: #666666">&</span>cntrl imin<span style="color: #666666">=1</span>, <span style="color: #666666">!</span> Minimize the initial structure maxcyc<span style="color: #666666">=10000</span>, <span style="color: #666666">!</span> Maximum number of cycles for minimization ncyc<span style="color: #666666">=5000</span>, <span style="color: #666666">!</span> Switch from steepest descent to conjugate gradient minimization after ncyc cycles ntb<span style="color: #666666">=1</span>, <span style="color: #666666">!</span> Constant volume ntp<span style="color: #666666">=0</span>, <span style="color: #666666">!</span> No pressure scaling ntf<span style="color: #666666">=1</span>, <span style="color: #666666">!</span> Complete force evaluation ntc<span style="color: #666666">=1</span>, <span style="color: #666666">!</span> No SHAKE ntpr<span style="color: #666666">=50</span>, <span style="color: #666666">!</span> Print to mdout every ntpr steps ntwr<span style="color: #666666">=2000</span>, <span style="color: #666666">!</span> <span style="color: #00A000">Write</span> a restart file every ntwr steps cut<span style="color: #666666">=10.0</span>, <span style="color: #666666">!</span> Nonbonded cutoff <span style="color: #AA22FF; font-weight: bold">in</span> Angstroms <span style="color: #666666">/</span> </pre></div> </td></tr></table>使用以下命令来完成能量最小化:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">pmemd</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 01_Min.in <span style="color:#666">-o</span> 01_Min.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> DOPC_128.inpcrd <span style="color:#666">-r</span> 01_Min.rst & </pre></div>能量最小化之后, 你可以读取.mdout文件来寻求最小化的细节. 进行下一步模拟的重要文件是01_Min.rst文件, 加热过程需要这个文件.
128个DOPC脂双分子层的能量最小化后的结构

初始的能量最小化之后, 我们将缓慢的将系统加热到预定温度. 在脂类双分子层模拟中, 选择合适的温度是非常重要的. 对于脂类双分子层的相变温度有很多可供参考的试验数据, 但是在模拟中重现其相变是一个困难的问题.
对于DOPC来说, 303K是一个合适的温度, 因为这个温度比相变温度高很多. 加热过程分为两步, 第一步将系统加热到100 K, 然后在保持系统结构同时将其缓慢加热到预定温度.
第一步加热使用如下输入文件02_Heat.in
注意初始加热使用了Langevin热浴, 以及nmropt=1允许你在加热过程中给体系用不同的温度. 脂类分子使用简谐限制来固定位置. 然后对于具体的GROUP输入选项在文件的末尾.
使用以下命令来完成第一步加热过程
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 02_Heat.in <span style="color:#666">-o</span> 02_heat.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 01_Min.rst <span style="color:#666">-r</span> 02_Heat.rst <span style="color:#666">-ref</span> 01_Min.rst <span style="color:#666">-x</span> 02_Heat.nc </pre></div>加热过程的第二步缓慢的将温度提升到预定温度. 这一步中, 原子的位置和速度从之前的重启(.rst)文件中读取. 这一次除使用Langevin方法调节温度, 还使用各向异性的Berendsen弱耦合方式调控压强.
使用如下命令来完成第二步加热过程
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">pmemd</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 03_Heat2.in <span style="color:#666">-o</span> 03_Heat2.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 02_Heat.rst <span style="color:#666">-r</span> 03_Heat.rst <span style="color:#666">-ref</span> 02_Heat.rst <span style="color:#666">-x</span> 03_Heat.nc </pre></div>128个DOPC脂双分子层加热后的结构
为了平衡周期性边界条件的系统, 如果使用GPU代码(pmemd.cuda), 先进行恒压的5 ns MD模拟是很重要的. 在正式使用MD模拟之前, 系统的密度和大小必须达到平衡. 因为周期性边界盒子尺寸一直在改变, 在500 ns模拟之后提高skinnb值是很重要的. 这样做可以避免大部分"skinny"报错.
我们这样做的原因是为了提高性能, GPU代码在模拟中不会重复计算非键的cells列表. 如果这些cells的尺寸改变了, 程序有很大概率出现skinnb相关的问题. 系统平衡之后会很大程度上稳定盒子尺寸, 从而避免报错.
我们将进行十次(5 ns)固定模拟来平衡系统.
使用以下命令来运行
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_1.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 03_Heat2.rst <span style="color:#666">-r</span> 04_Hold_1.rst <span style="color:#666">-x</span> 04_Hold_1.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_2.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_1.rst <span style="color:#666">-r</span> 04_Hold_2.rst <span style="color:#666">-x</span> 04_Hold_2.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_3.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_2.rst <span style="color:#666">-r</span> 04_Hold_3.rst <span style="color:#666">-x</span> 04_Hold_3.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_4.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_3.rst <span style="color:#666">-r</span> 04_Hold_4.rst <span style="color:#666">-x</span> 04_Hold_4.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_5.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_4.rst <span style="color:#666">-r</span> 04_Hold_5.rst <span style="color:#666">-x</span> 04_Hold_5.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_6.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_5.rst <span style="color:#666">-r</span> 04_Hold_6.rst <span style="color:#666">-x</span> 04_Hold_6.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_7.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_6.rst <span style="color:#666">-r</span> 04_Hold_7.rst <span style="color:#666">-x</span> 04_Hold_7.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_8.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_7.rst <span style="color:#666">-r</span> 04_Hold_8.rst <span style="color:#666">-x</span> 04_Hold_8.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_9.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_8.rst <span style="color:#666">-r</span> 04_Hold_9.rst <span style="color:#666">-x</span> 04_Hold_9.nc <span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 04_Hold.in <span style="color:#666">-o</span> 04_Hold_10.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_9.rst <span style="color:#666">-r</span> 04_Hold_10.rst <span style="color:#666">-x</span> 04_Hold_10.nc </pre></div>我们可以使用以下文件来进行实际模拟. 这里控温使用了Langevin热浴, 控压使用了各向异性的Berendsen控压法.
使用此文件作为输入05_Prod.in
使用如下命令来成品模拟
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">pmemd.cuda</span> <span style="color:#666">-O</span> <span style="color:#666">-i</span> 05_Prod.in <span style="color:#666">-o</span> 05_Prod.out <span style="color:#666">-p</span> DOPC_128.prmtop <span style="color:#666">-c</span> 04_Hold_10.rst <span style="color:#666">-inf</span> <span style="color:#666">-r</span> 05_Prod.rst <span style="color:#666">-x</span> 05_Prod.nc </pre></div>128个DOPC脂双分子层5 ns成品模拟后的结构

使用重启文件和相同的输入文件, 此模拟可以持续到结构收敛为止. 特别的, 通常要将成品模拟继续进行, 直至结构中每个脂分子的面积收敛.
cpptraj分析在模拟完成后, 有很多值得分析的地方, 比如单个脂类分子表面积, 电荷密度, 氘代序参数等等. 当下文献中模拟软件提供很多分析的方法, 在此简单介绍.
有很多脚本可供分析脂类双分子层结构. 本章将为读者解释如何从轨迹中得到每个脂类分子的面积以及平均电荷密度.
脂类分子面积一般指代单分子平均面积, 用平方埃表示.
单个脂分子平均面积=(盒子X方向尺寸)*(盒子Y方向尺寸)/(每层的脂类数目)
cpptraj是Amber自带的轨迹分析程序. 为了计算双分子系统中每个脂类分子的面积, 你可以使用这个程序来从轨迹中提取盒子尺寸信息. 在运行cpptraj之前, 我们需要一个输入文件. 以下给出一个样本.
使用以下命令运行cpptraj:
vector.dat文件中储存了盒子的尺寸信息, 但是文件的前两行是头文件, 在计算中需要删除掉.
用tail命令来去除前两行
为了计算分子面积和时间的关系, x方向尺寸(第二列)需要和Y方向尺寸(第三列)相乘.
使用awk命令来实现
area_per_lipid.dat可以简单的用绘图软件画出. 比如, 使用gnuplot绘图, 首先打开gnuplot.
作图
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">plot</span> "area_per_lipid.dat" with lines </pre></div>128个DOPC脂分子双膜中每个脂分子的面积
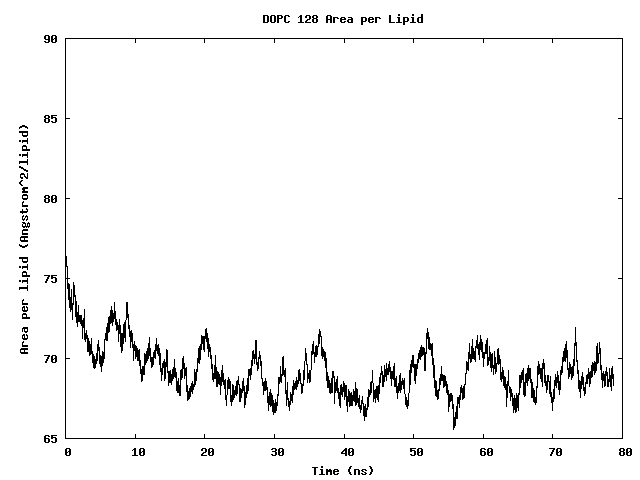
可以看到经过一定时间的模拟之后, 每个脂分子的面积收敛了.
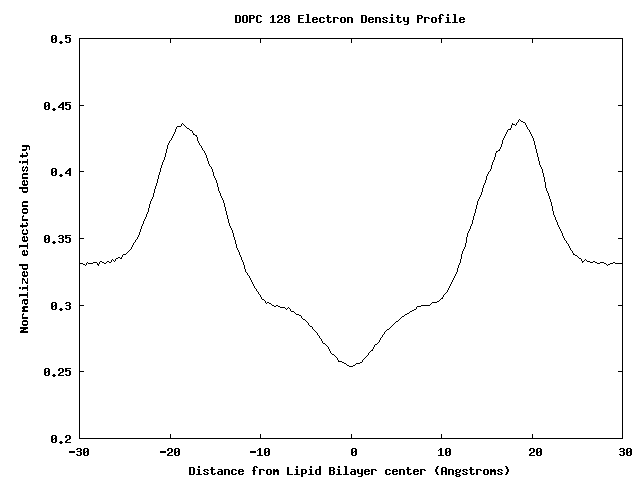
电荷密度信息提供给我们一段时间内电荷的平均密度. 这可以从轨迹文件中直接的计算得到. 电荷密度图像经常被拿来与实验模型的电荷密度比对.
用于分析脂类的函数已经被加入了cpptraj程序. cpptraj现在已经可以计算电荷密度函数.
为了使用cpptraj来计算电荷密度, 和上一节一样, 我们依旧需要输入脚本文件.
以下给出样本density.cpptraj
使用如下命令来运行修改后的ptraj执行脚本:
现在, 我们有了包含电荷密度的文件, electron_density.dat
你可以使用你习惯的程序来绘图, 以下给出gnuplot的例子:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">gnuplot</span> <span style="color:#A2F">plot</span> "electron_density_normalized.dat" using 1:2 with lines </pre></div>128个DOPC脂分子双膜的电荷密度剖面
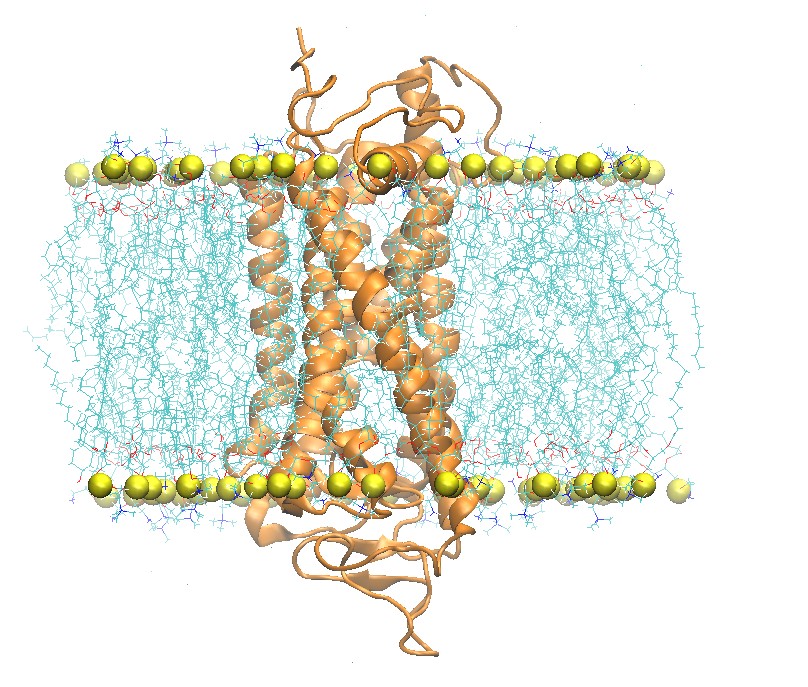
2:2:1 POPC:POPE:Cholesterol脂分子双层膜中的视紫红质
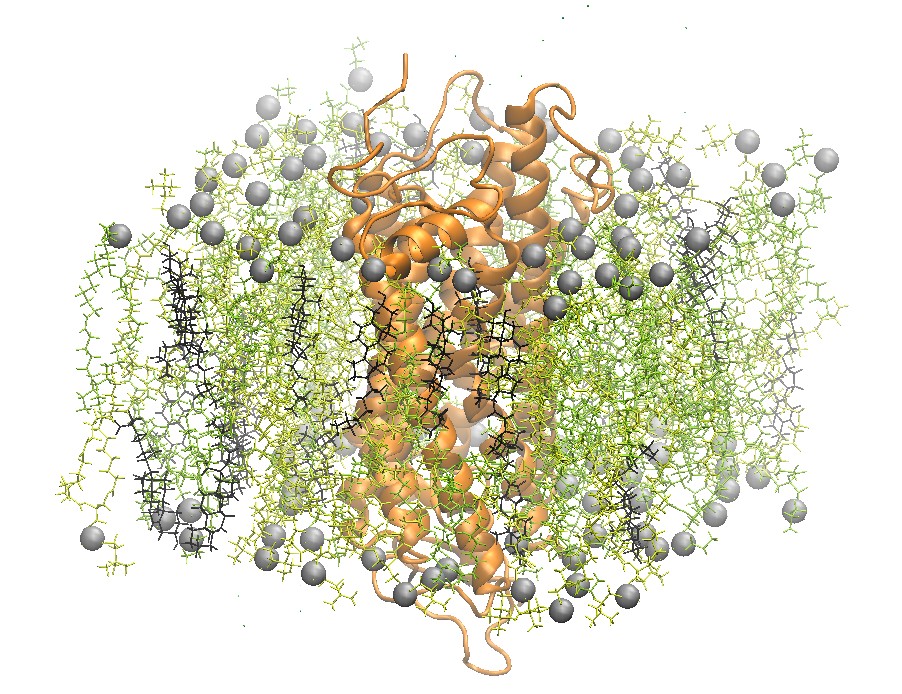
一个最近研究的膜结合蛋白质体系是视紫红质, 感光细胞中的视觉色素. 视紫红质是最早被结晶的G蛋白偶联受体. Grossfield团队使用Blue Gene超算, 当时世界上的领先超算, 在相当长的时间尺度下对视紫红质进行了研究. 特别是他们得以验证受体中的水分子动力学以及水分子对视紫红质的影响[5].
在这一节里, 我们列出如何构建结构以及进行模拟的提纲.
视紫红质Rhodopsin(1U19)的晶体结构, 包含水分子氧和Retinal
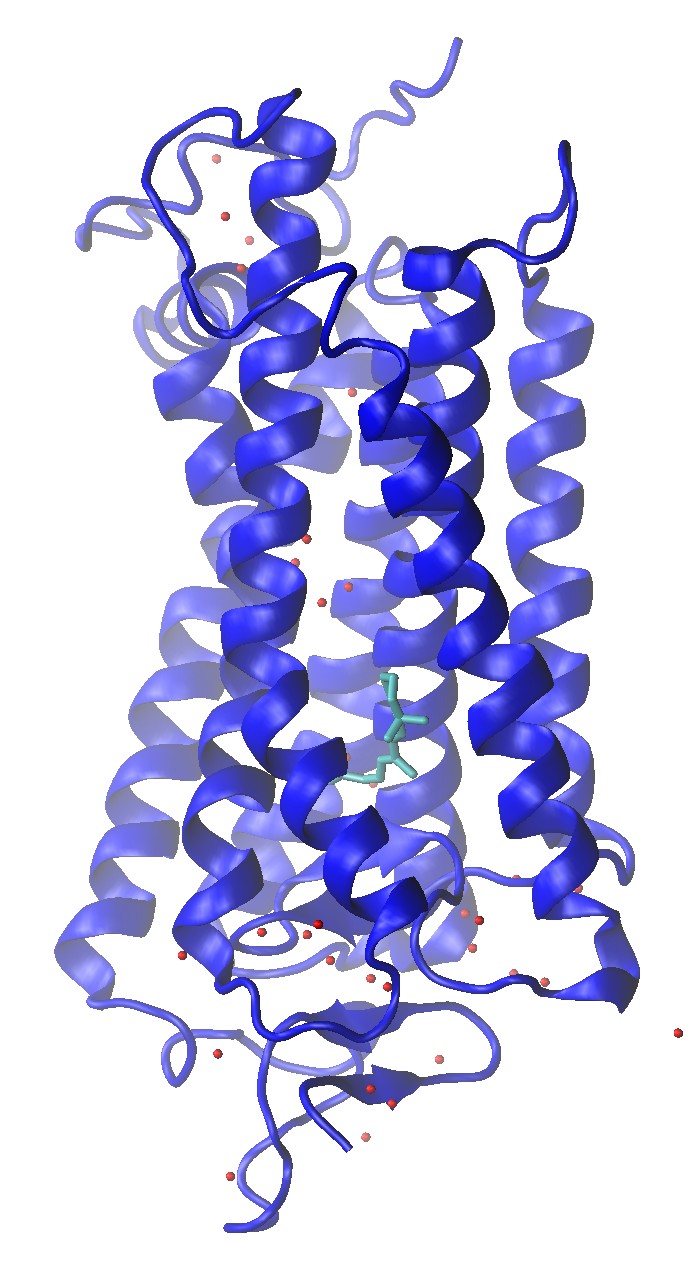
视紫红质的晶体结构可以在RCSB PDB数据库中找到, 编号1U19.
对于此系统, 我们可以再次使用CHARMM-GUI来搭建结构.
第一步: 选择Protein/Membrane System, 输入RCSB PDB ID 1U19, 然后选择RCSB作为结构类型.
第二步: 仅保留A链作为初始结构, 继续下一步.
第三步: 继续完成PDB文件, ACE残基必须被重命名以从结构中移除, 在Cysteine残基的110到187残基中需要有二硫键
第四步: 选择Align the First Principle Axis Along Z选项
第五步: 计算横截面积和系统体积, 选择50:50:25 POPC:POPE:Cholesterol的比例
第六步: 将离子浓度设置为0.07 M KCL
之后使用默认设置直到第五步完成, 在此之后, 系统已经组装好, 可以下载了.
step5_assembly.pdb结构图如下
2:2:1 POPC:POPE:Cholesterol脂分子双层膜中视紫红质的顶视图和侧视图
此pdb文件可以用charmmlipid2amber.py处理
警告: 在此例中, CHARMM-GUI在蛋白链后不包括TER. 如果包含的话, 在用charmmlipid2amber.py处理之前一定要将TER结束标签加入到PDB文件中.
之后就是用我们熟悉的LEaP载入了. 在LEaP中可以载入力场, 如ff12SB结合Lipid14力场, 然后就可以构建拓扑文件了.
[1] Dickson, C.J., Madej, B.D., Skjevik, A.A., Betz, R.M., Teigen, K., Gould, I.R., Walker, R.C., "Lipid14: The Amber Lipid Force Field", J. Chem. Theory Comput., 2014, 10(2), 865-879. DOI: 10.1021/ct4010307 [2] Skjevik, A.A,; Madej. B.D.; Walker, R.C.; Teigen, K.; "LIPID11: A Modular Framework for Lipid Simulations Using Amber", Journal of Physical Chemistry B, 2012, 116 (36), pp 11124-11136. DOI: 10.1021/jp3059992 [3] Dickson, C.J.; Rosso, L.; Betz, R.M.; Walker, R.C.; Gould, I.R., "GAFFlipid: a General Amber Force Field for the accurate molecular dynamics simulation of phospholipid.", Soft Matter, 2012, 8, 9617-9627. DOI: 10.1039/C2SM26007G [4] Kucerka, N.; Tristram-Nagle, S; Nagle, J. J. Membrane Biol. 2005, 208, 193-202. [5] Alan Grossfield, Michael C. Pitman, Scott E. Feller, Olivier Soubias, Klaus Gawrisch, Internal Hydration Increases during Activation of the G-Protein-Coupled Receptor Rhodopsin, Journal of Molecular Biology, Volume 381, Issue 2, 29 August 2008, Pages 478-486, ISSN 0022-2836.