<em>Amber</em>是一套让用户能够进行分子动力学模拟的程序组, 特别是用于生物分子的模拟. 没有单个的程序使用<em>Amber</em>这个名称, 但各部分能很好地协同工作, 并为许多常用的计算提供了强大的框架. [1, 2] 术语<em>Amber</em>也用来指实现的经验力场. [3, 4]然而, 应当认识到, 该代码和力场是分开的: 若干其他计算程序包已经实现了<em>Amber</em>力场, 并且<em>Amber</em>程序组也可以使用其他力场. 此外, 该力场是公开的, 而程序代码需要授权许可协议.
Amber软件组分为两部分: AmberTools17, 一组可免费获取的程序组, 多数遵循GPL证书; Amber16, 以pmemd模拟程序为中心, 除继续以前的证书协议外, 还受更有约束力的证书限制. 相比最近的以前版本Amber14, Amber16 代表了一个显著的变化(我们将版本编号移到了年份的最后两个数字上, 因此没有版本13或15). 请参阅http://ambermd.org上重要变化的概述.
AmberTools 是一套用于生物分子模拟和分析的程序. 它们设计用于互相配合, 协同工作, 也同样与"常规" Amber 程序组协同工作. 您可以使用 AmberTools 执行许多模拟任务, 您可以使用 AmberTools 和 Amber 本身的组合进行更广泛的模拟. AmberTools 的大部分组件都是在GNU通用公共许可证(GPL)下发布的. 少量组件属于公共领域或拥有其他开源许可证. 请参阅<em>README</em>文件获取更多信息.
每个人都应该阅读(或至少略读)本章. 即使你是一位经验丰富的Amber用户, 这里仍可能有一些你忽略了的, 或全新的功能, 这些是有帮助的. 在Amber网站http://ambermd.org上也有提示和例子. 虽然Amber在刚出现时可能极为复杂, 但在过去的几年中, 使用已经变得更容易, 一旦你理解了基本架构和选项的选择, 总体来说就相当简单了. 特别是, 我们尽力编写的教程, 使新用户也能使用它. 成千上万的人已经学会了使用Amber; 不要轻易气馁.
如果您想了解更多关于基本生化模拟的技术, 有很多好书可以参考, 从入门的描述, [5-7]到液态模拟的标准方法, [8-10]到很多作者合编的涵盖很多生物分子建模的重要方面的书籍. [11-15]寻找和你可能想进行的模拟相似的论文"范例", 通常也是一个好主意. 如果您是这个领域的新手, 第14章提供了力场的基本介绍, 以及在Amber文件中编码参数的细节.
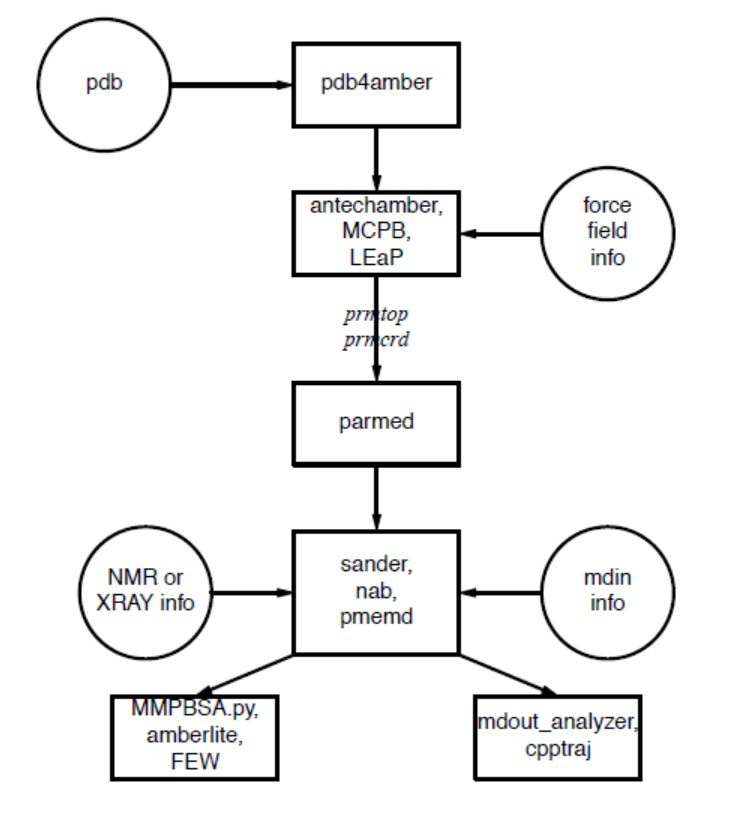
理解 AmberTools 从哪开始是管理这个包中信息流的主要问题-图1.1. 您首先需要了解模拟程序需要什么信息(sander, pmemd, mdgx或nab). 您需要知道它来自哪, 以及它如何进入这些程序所需要的形式. 本部分旨在指导新用户, 而不是替代单个程序文件.
图1.1 Amber的基本信息流
所有模拟程序需要的信息(见图1.1中的圆圈):
系统中每个原子的笛卡尔坐标. 它们通常来自X-射线晶体学, NMR谱或建立的模型. 它们通常应该是蛋白质数据库(PDB)格式. LEaP程序为执行许多建模任务提供了一个平台, 但用户也不妨考虑一下其他程序. 一般来说, 需要编辑这些文件, pdb4amber脚本可以做一些工作.
拓扑: 连接性, 原子名称, 原子类型, 残基名称和电荷. 这个信息来自于数据库, 位于$AMBERHOME/dat/leap/lib目录中, 并在第3章中介绍. 它包含标准氨基酸以及N-和C-末端带电氨基酸, DNA, RNA, 以及常见的糖和脂质的拓扑结构. 其他分子的拓扑信息(未在标准数据库中找到)保存在用户生成的"残基文件"中, 这些文件通常是使用antechamber创建的.
力场: 系统中所有键, 角, 二面角和原子类型的参数. 几种力场的标准参数可以在$AMBERHOME/dat/leap/parm目录中找到; 更多信息见第3章. 这些文件可以"按原样"用于蛋白质和核酸, 用户也可以准备它们自己的文件 其中包含对标准力场的修改.
一旦创建了拓扑和坐标文件(通常称为prmtop和prmcrd, 但是也可以使用任何合法的文件名), parmed脚本可以用来检查和验证它们, 并进行修改. 特别是, checkValidity操作将会标记许多潜在的问题.
命令: 用户指定所需的程序选项和状态参数. 这些在输入文件(默认名为mdin)或在用NAB语言编写的"驱动程序"程序中指定.
LEaP是在Amber中创建一个新系统或修改现有系统的主要程序. 它可以作为tleap或图像界面xleap的命令行程序. 它结合了Amber早期版本的prep, link, edit和parm功能.
pdb4amber通常有助于将其他地方(如https://rcsb.org)创建的PDB格式的文件与LEaP兼容
parmed提供了一种简单的从参数拓扑文件中提取参数定义信息的方法. 它也可以用来检查参数拓扑文件对于复杂系统是否有效(请参阅checkValidity命令), 它也可以快速地对这个文件进行简单的修改.
antechamber是发展药物分子或改性氨基酸使用的一般Amber力场(GAFF)的主要程序. 这些可以直接在LEaP中使用, 也可以作为进一步参数开发的新出发点.
MCPB.py提供了一种建立, 原型和验证金属蛋白和有机金属化合物MM模型的方式. 它使用静电键合模型来扩展现有的成对累加力场. 这是一个在Python中重新实现MCPB, 从以前的版本自动合并的具有更高效的工作流程和建模过程.
IPMach.py提供了一个方便的离子非键合模型(12-6 LJ模型和12-6-4LJ型)参数化的工具.
paramfit允许通过拟合量化数据来生成任何分子的键合力场参数.
sander(AmberTools的一部分)是能量最小化和分子动力学的基本程序. 这个程序通过迭代将原子沿能量梯度移动到足够低的平均值来放松该结构. 分子动力学部分通过集合牛顿运动方程来产生系统的构象. MD采样将得到比最小化更多的构象, 并会允许结构跨越小的势能势垒. 在模拟过程中, 构象可以定期保存, 用于以后的分析, 并使用热力学积分进行基本自由能计算. 更精细的构象搜索和建模MD研究也可以使用sander模块进行. 这允许将多种约束条件添加到基本力场中, 并且已经被专门设计用于计算包括NMR, X射线或冷冻EM结构细化.
pmemd(Amber的一部分)是针对速度和平行缩放而优化的sander版本; pmemd.cuda是在GPU上运行的变种. 这个名字代表"Particle Mesh Ewald Molecular Dynamics", 但是这个代码可以现在也进行广义Born模拟. 在sander上输入和输出只有少量变化.
mdgx是一个分子动力学工具, 模仿了sandder和pmemd中的一些功能, 但具有简单的C代码和原子排序程序, 可以简化力计算过程中的信息流. mdgx的主要目的是提供重新设计基本分子动力学的工具算法和模型, 并支持参数开发的新模型.
NAB(Nucleic Acid Builder)是一种可用于编写程序来执行非周期性模拟的语言, 经常用于隐式溶剂力场.
mdout_analyzer.py是一个简单的Python脚本, 它将提供来自sander或pmemd的输出文件中的信息摘要.
cpptraj是用于进行叠加, 提取坐标, 键/角/二面角值计算, 原子位置波动, 相关函数, 氢键分析等的主要轨迹分析工具(用C ++编写). 更多信息见第28章.
pytraj是Python包装的cpptraj. 通过与Python丰富的echosystems(如numpy, scipy, ipython-notebook ...)相结合, 在数据分析中引入更多的灵活性.
pbsa是溶剂介导的生物分子能量分析程序. pbsa.cuda是在 GPU上运行的变体. 它可以用来静电和非静电连续溶剂化计算, 用于来自分子动力学模拟和其他来源(以pqr格式)的输入坐标文件. 它还支持在各种可视化程序中对溶剂介导的静电势进行可视化. 更多信息见第6章.
MMPBSA.py是一个可以使用连续溶剂模型生成的思想自动进行分子动力学模拟的能量分析python脚本. (还有一个老版本的Perl脚本, 名为mm_pbsa.pl, 具有类似的功能. )
FEW(自由能工作流)使用TI, MM/PBSAtype或LIE自动计算蛋白质-配体结合的自由能.
amberlite是一组NAB程序和python脚本, 实现有限的MD模拟和mm-pbsa(或mm-gbsa)分析, 主要针对蛋白质-配体相互作用的分析. 这些工具本身也是可以用的, 也可以很好地引入Amber中, 也可以作为更复杂计算的起点.
Xtal分析一系列用于分析晶体仿真轨迹的工具. 更多信息见第33章.
<em>Amber</em>由大量程序组成, 旨在帮助您对化学系统进行计算研究, 并且发布工具的数量也会不断增长. 本节列出了主要程序包括AmberTools. 程序组包含的每个程序都在这里列出, 简要描述了它的主要功能以及手册中的哪一章可以找到更全面的描述. 注意: 有一些额外的程序, MTK++组的一部分, 这里没有列出. 请参阅它的文档以获取更多信息.
AddToBox: 添加溶剂分子到晶胞的程序. 见16.3部分.
CartHess2FC.py: 使用Seminario方法基于Cartesian Hessian矩阵推导力常量的程序. 见15.8.2.5部分.
car_to_files.py: 基于 car文件生成mol2和PDB文件的程序. 见15.8.2.8部分.
ChBox: 用于更改Amber盒子尺寸的启动程序. 见16.4部分.
CheckMD: 用于自动检查MD模拟的程序. 运行没有选择声明的程序.
IPMach.py: 一个用于提取非键合离子模型参数的程序. 见15.8.2.2部分.
MCPB.py: MCPB的优化python版本. 见15.8.2.1部分.
MMPBSA.py: 根据MM/PBSA近似计算结合自由能用于处理后轨迹的程序. 见第30章.
MTKppConstants: 列出MTK++中使用的常量. 运行没有参数的程序来获取完整列表.
OptC4.py: 优化在蛋白质系统中金属位点复合物的C4项. 见15.8.2.4部分.
PdbSearcher.py: MTK++程序中Pdbsearcher的python版本. 见15.8.2.3部分.
PropPDB: 扩增PDB结构的程序. 见16.2部分.
ProScrs.py: 用于将蛋白质片段切割和封盖成簇的程序. 见15.8.2.7部分.
UnitCell: 从PDB结构重新创建晶体晶胞的程序. 见16.17部分.
Acdoctor: 一个诊断什么可能导致antechamber失败的工具. 见15.5.1部分.
am1bcc: antechamber中用于在配体参数化过程中计算AM1-BCC电荷的调用程序. 它可以作为一个独立的程序使用, 当您输入没有参数的程序名时的时候. 见15.4部分.
Ambpdb: 将Amber系统(prmtop和inpcrd/restart)转换为PDB, MOL2或PQR文件的程序. 参见27.1部分.
ante-MMPBSA.py: 用于为具有单启动拓扑文件的MMPBSA创建必要的、自洽的prmtop文件的程序. 见30.2.2部分.
antechamber: 用于配体和其他小分子参数化的程序. 见第15章
atomtype antechamber: 中用于判断输入结构中原子类型的调用程序. 它可以作为一个独立的程序使用. 如果您不提供参数, 它会打印出用法语句. 见15.4节部分.
Bondtype: antechamber中用于判断给定输入结构中存在哪些类型的键的调用程序. 它可以作为一个独立的程序使用. 如果您不提供参数, 它会打印出用法语句. 见15.4部分.
charmmlipid2amber.py: 将使用CHARMM-GUI脂质生成器创建的PDB转换为能被Amber和AmberTools程序识别的PDB的脚本. 如果您不提供参数, 则会打印出用法语句.
cpinutil.py: 从PDB文件创建一个恒定的pH输入(CPin)文件的程序. 如果您不提供参数, 你会得到使用说明.
cpptraj: 用于处理后轨迹和数据分析的多功能程序. 见第28章.
cphstats: 计算恒定pH模拟下的质子化状态统计的程序. 见22.7部分.
elsize: 估计一个给定的输入结构的有效静电大小的程序. 见4.2.1部分.
espgen: antechamber中用于在配体或小分子参数化期间生成ESP文件的调用程序. 它可以作为一个独立的程序使用. 如果您不提供参数, 它会打印出用法语句.
espgen.py: espgen的python版本. 见15.8.2.6部分.
ffgbsa: 作为amberlite包的一部分, 计算MM/GBSA能量的程序.
FEW.pl: 自动计算自由能工作流的程序. 见第32章
gbnsr6: 计算基于广义Born的溶剂接触表面积自由能的程序. 见第5节
hcp_getpdb: 向拓扑(prmtop)文件添加必要的部分, 以便用于HCP GB近似的程序. 见第39.6节
makeANG_RST: 为sander的 nmropt = 1功能设置角度约束的程序.
makeCHIR_RST: 为sander的 nmropt = 1功能设置手性约束文件的程序.
makeDIP_RST.cyana: 为sander的 nmropt = 1功能基于CYANA的偶极子信息进行限制的程序.
makeDIST_RST: 为sander的 nmropt = 1功能设置距离限制的程序.
matextract: 对称定义程序的一部分, 用于从标准输入转储到标准输出的打印矩阵. 见第37.5.5节
matgen: 生成对称转换矩阵. 对称定义程序的一部分. 见第37.5.1节
matmerge: 将对称变换矩阵合并成一个矩阵变换矩阵. 对称定义程序的一部分. 见第37.5.3小节
matmul: 将矩阵相乘. 对称定义程序的一部分. 见第37.5.4小节
mdgx: 显式溶剂分子动力学引擎, PME. 见第26章
mdnab: 作为Amberlite的一部分在NAB中编写的隐性溶剂MD程序, 见第41章
mdout_analyzer.py: 一个允许您快速分析和来自sander/pmemd输出文件的图形数据的脚本. 见第27节
minab: 作为Amberlite的一部分在NAB中编写的隐式溶剂最小化程序, 见第41章
mm_pbsa.pl: 用于执行MM/PBSA计算的较早的perl脚本. 鼓励新用户改用MMPBSA.py.
mm_pbsa_statistics.pl: mm_pbsa.pl的补充脚本, 用于从完整的mm_pbsa计算中计算MM/PBSA统计信息.
mm_pbsa_nabnmode: 通过mm_pbsa.pl对生物分子进行最小化和正常模式分析的程序.
mmpbsa_py_energy: 一个编写的NAB程序, 用于计算使用GB或PB溶剂模型MMPBSA的能量. 它可以作为一个独立的程序来模仿sander的imin = 5功能, 但它是在MMPBSA中自动调用. 请参阅MMPBSA mdin文件作为此程序的示例输入文件. 提供-help或-h标志打印使用消息.
mmpbsa_py_nabnmode: 一个编写的NAB程序, 用于计算MMPBSA的正常模式熵贡献, 这只能由MMPBSA使用.
Molsurf: 基于输入PQR文件和探针半径计算分子表面积的程序. 不提供参数打印使用信息.
NAB: 代表核酸生成器. NAB实际上提供了一个大体上基于C的方便的分子编程语言的编译器. 见第35章和其他相关章节.
new_crd_to_dyn: 从坐标文件中设置Tinker风格的.DYN文件. 这是Tinker-to-Amber转换程序的一部分.
new_to_old_crd: 将新的Tinker风格坐标文件转换为旧的Amber风格坐标. 这是Tinker-to-Amber转换程序的一部分.
Nmode: 一个用于计算生物分子正常模式的过时程序. 鼓励您使用NAB代替. 见第39.1节
paramfit: 通过拟合量子数据来改善力场参数. 见第11章
parmcal: 交互式计算给定角度和键的参数. 见15.5.2部分
parmchk2: 一个分析输入力场库文件(mol2或amber prep)的程序, 并提取相关的参数写入frcmod文件. 见15.1.2部分
parmed: 查询和操作prmtop文件的程序. 见14.2部分
pbsa: 一个计算静电和非静电连续溶剂化自由能的程序. 见第6章
pbsa.cuda: 一种GPU加速版的pbsa. 见第6章
pdb4amber: 一个leap中用于准备PDB文件的程序. 见12.4部分
pmemd: 一个性能和并行优化的动态引擎, 实现了一个sander的部分功能.
pmemd.cuda: 一个GPU加速版pmemd
prepgen: antechamber中用作生成Amber prep文件的程序, 不提供参数打印使用信息. 见15.4部分
pymdpbsa: 对MD(GB, PB)/ SA计算完整分析的工具. 见第41.7部分
pytleap: 用于leap 和antechamber运行为Amber准备拓扑和坐标文件的用户容易掌握使用的包装. 见第41.3部分
pytraj: 一个结合cpptraj的Python程序. 见第29节
reduce: 向PDB添加或去除氢原子的程序. 见12.5部分
residualgen: 一个用于自动生成Amber残基模板的程序(即Amber prep文件). 见15.5.3部分
resp: 通常由antechamber和R.E.D工具调用的程序, 用于执行静电约束来计算部分原子电荷. 不提供参数打印使用信息
respgen: 一个antechamber调用用于生成RESP输入文件的程序. 见15.4部分
rism1d: 1D-RISM求解程序. 见7.4部分
rism3d.snglpnt: 用于单点计算的3D-RISM求解程序. 见7.6部分
sander: 用于运行Amber分子模拟的主要引擎. 最初是用核磁共振能量约束模拟退火的缩写.
saxs_rism: 从3D-RISM输出计算小(大)角X射线散射曲线的程序
saxs_md: 从MD轨迹计算小(大)角X射线散射曲线的程序
softcore_setup.py: 一个帮助软核TI设置sander的程序. 不提供参数获得使用信息.
sqm: 半经验(或独立)量子力学求解程序. 见第9章
tinker_to_amber: 将Tinker analout和参数文件转换为Amber兼容的拓扑文件.
tleap: 调用teLeap用于设置特定命令行参数的脚本. 见第13章
transform: 将矩阵变换应用于结构. 对称定义程序的一部分. 见37.5.6部分
tss_init: 一个做一些有关矩阵的程序. 见第37.5节
tss_main: 一个做一些有关矩阵的程序. 见第37.5节
tss_next: 一个做一些有关矩阵的程序. 见第37.5节
ucpp: 一个做一些源代码预处理的程序. 你永远不应该使用这个程序 - 它被nab使用.
xaLeap: 一个用于创建Amber拓扑文件的图形程序. 这个程序是通过xleap脚本调用的, 所以你不应该直接调用这个程序.
xleap: 调用xaLeap用于设置特定命令行参数的脚本. 见第13章
xparmed: ParmEd功能的图形前端(即参数文件编辑和查询). 见14.2部分
本章将概述如何安装和测试您的版本. Amber网页(http://ambermd.org)有各种常见操作系统的附加说明和提示. 寻找"Running Amber on ...."链接. 下载完版本文件后, 请执行以下操作:
/home/myname作为例子):确认将上面的/home/myname更改为适合您的机器的任何目录, 并且确保您在您选择的目录树中拥有写入权限.
更多有关信息, 请参阅http://ambermd.org/amber_install.html以及其他类型的Linux和Macintosh OSX的要求.
会为您显示选项. 选择你想要的编译器和标志; 对于大多数系统来说, 应该如下操作:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">./configure</span> gnu </pre></div>此步骤还将检查是否有任何更新和错误修复尚未应用于您的安装, 并将应用它们(除非你不要). 如果配置步骤找到丢失的库, 请回到步骤3. 这一步也会询问你是否要为Python安装在Amber中兼容的Python可执行文件程序包括(MMPBSA.py, MCPB.py, ParmEd, pysander, pytraj, pdb4amber, 其余的amberlite). 由于Amber现在需要Python 2.7或更高版本, 以及numpy, scipy和matplotlib启用其所有的功能, 现在配置提供了一个选项来下载兼容的PythonContinuum IO(通过miniconda)安装在Amber目录中, 用于Amber程序. 更多细节见2.2部分. 只要你的默认Python已经安装了必要的条件, configure就会只需选择与Amber一起使用的Python. 不要在这一步选择任何并行选项! 您需要先安装串行版本; 在步骤8下面描述了用于并行的构建. 要自动接受配置过程中的所有建议, 可以将"yes"的输出传送给配置:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">yes</span> | ./configure gnu </pre></div>这些源代码脚本将为Amber正确设置您的shell环境:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">source</span> /home/myname/amber16/amber.sh <span style="color:#080;font-style:italic"> #(对于bash, zsh, ksh等)</span> <span style="color:#A2F">source</span> /home/myname/amber16/amber.csh<span style="color:#080;font-style:italic"> #(对于csh, tcsh)</span> </pre></div>当然, /home/myname/amber16应该针对你的AMBERHOME进行调整. 添加这些命令到您的系统资源文件(例如, ~/.bashrc, ~/.cshrc, ~/.zshrc等)将会在你开启一个新的shell时, 自动设置您的环境. 注意, 这一步对于运行Amber中任何Python模块都是绝对必要.
将编译代码. 如果此步骤失败, 请尝试仔细阅读错误消息以确定问题.
将运行测试, 并报告成功或失败. 在发现"possible FAILURE"消息时, 转到 $AMBERHOME/AmberTools/test下的指定目录或$AMBERHOME/test, 然后查看"*.dif"文件. 差异可能涉及到最后一位数字的四舍五入印刷的或偶尔因机器而异的信息(详见下文). 和汇编一样, 如果您在单个测试中遇到麻烦, 您可能选择在Makefiles中注释某些行(即$AMBERHOME/AmberTools/test/Makefile或$AMBERHOME/test/Makefile), 和直接进入测试子目录详细检查输入和输出. 为了方便, 所有的错误信息和差异收集在$AMBERHOME/logs目录中;如果除了舍入误差外, 还有其他的东西, 你可以快速地察看它们.
分子动力学的本质是计算的过程非常依赖于顺序算术运算和实现的机器算法, 即用于舍入的方法. 因为计算的每一步都取决于前一步的结果, 所以稍有不同都会导致最终轨迹的差异. 一个最初相同的动力学运行在两个不同的机器, 最终的轨迹将也可能变得完全不相关. 它们都不是错的;它们只是在探索相空间的不同区域. 因此, 很长的一段时间模拟后的状态对于验证正确性并不是很有用. 如果正常, 考虑到统计波动的平均值是有意义的. 在这种情况下, "不同的机器"意味着浮点硬件, 字大小或舍入模式, 以及编译器或库中的任何差异. 算术运算顺序的差异会影响舍入行为; (a + b)+ c不一定与a +(b + c)相同. 不同的优化级别会影响操作顺序, 因此可能会影响计算的过程.
所有整数的初始值应该是相同的. 第一个周期的能量和温度应该是相同的. 在sander中的RMS和MAX梯度通常比能量更精确, 并且在某些机器的最后一个图中可能会有1的变化. 在最小化和动力学计算中, 在小至100-200周期之后发现小的行为差异并不罕见.
注意: 如果您解压了Amber16.tar.bz2文件, 则步骤1-6将安装并测试AmberTools和Amber; 否则它只会安装并测试AmberTools. 如果您后来获得了Amber的授权, 请返回并重复步骤1-6.
这假定你已经安装了MPI, 而mpicc和mpif90在你的PATH中. 一些MPI安装被调整到特定的硬件(比如infiniband), 如果你有这样的硬件, 你应该使用这些版本. 大多数人可以使用mpich或openmpi的标准版本. 使用我们已经准备的简单的脚本安装其中之一:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">cd</span> $AMBERHOME/AmberTools/src <span style="color:#A2F">./configure_mpich</span> <compiler-choice> OR <span style="color:#A2F">./configure_openmpi</span> <compiler-choice> </pre></div>按照这些脚本的说明, 然后返回到步骤7开始.
Amber 16有一个额外的标志(-intelmpi), 以便使用英特尔MPI库. 用这个来代替步骤8中的-mpi, 要确保已经安装了英特尔MPI库以及该mpiicc和mpiifort在你的PATH中.
关于AmberTools中并行程序的一些注意事项:
MPI版本的nab被称为mpinab, 类似于mpicc或mpif90: mpinab是一个将NAB语言编写的源代码生成一个MPI启用的可执行文件的编译器. 在编译mpinab之前, 请确保您熟悉nab的串行版本, 并且您确实需要一个并行版本. 如果您有共享内存节点, 则OpenMP版本可能是更好的选择. 更多信息见第35.4节. (注意, mpinab主要用于编写调用MPI版本的能量函数驱动程序;它不需要编写你自己的, 新的, 并行代码. )
MMPBSA.py的MPI版本被称为MMPBSA.py.MPI, 并且需要运行mpi4py包. 如果它不在您的Python标准库中, 它将与MMPBSA.py.MPI一起构建并放入$AMBERHOME前缀. 如果你有关于MMPBSA.py.MPI的问题, 看看串行版本MMPBSA.py你是否遇到同样的问题, 以查看它是否是并行版本或MMPBSA.py的一般问题. 由于我们不作或维护mpi4py源代码, 因此MMPBSA.py.MPI将在没有构建mpi4py时平台不可用.
NAB和Cpptraj也可以使用OpenMP进行编译:
注意, NAB的OpenMP版本与单线程版本具有相同的名称. 有关运行OpenMP版本的NAB的信息, 参见第35.4节;有关运行OpenMP版本的Cpptraj的相关信息, 参见第28.1.6.2节.
有关安装GPU加速版本的pmemd的信息, 请参见第18.6.5节.
关于安装GPU加速版本的pbsa的信息, 请参见第6.6.4节
所有Amber和AmberTools都包含在$AMBERHOME目录中. 所以删除这个目录会从您的计算机上彻底删除Amber. 但是, 有些情况下, 您可能希望删除一些编译Amber时创建的文件和程序;本节描述了为了这些目的而提供的Makefile规则.
make clean: 此命令将删除编译Amber时创建的所有临时对象文件(这些文件通常位于与源代码相同的目录中). 例如, 当你打算构建一个新的Amber程序变体(比如构建OpenMP并行程序或MPI并行程序)时是必要的. 这由配置脚本自动完成. 如果你通过手动编辑config.h文件或者设置AMBERBUILDFLAGS环境变量来修改编译器标志, 这就有必要了. 这些被认为是高级选项.
make distclean: 这个命令是一个大锤 - 它删除所有的临时文件, 已安装的库和程序, 以及几个第三方库(但不可能是你已经单独安装的东西, 像通过configure_openmpi或configure_mpich脚本实现的MPI). 这个命令的目的是将源代码树返回到原始状态, 就好像您刚刚提取了一个新版本的源代码. 如果您计划更改您用于构建Amber的编译器, 你应该先运行这个命令.
注意, 以上任何命令都不会逆转您已经应用的任何更新, 它们仅影响在安装过程中创建的文件和程序.
最近Amber的发展已经更多地将Python编程语言作为其几个组件的核心语言. 除了像MMPBSA.py, MCPB.py和ParmEd这样的独立程序之外, 越来越多的组件也通过Python API(如pysander, ParmEd和pytraj)表达了Amber的大量功能.
Amber的Python程序和库充分利用了基于numpy数值编程包的丰富, 成熟和强大的科学计算生态系统. 许多Amber Python包的标准依赖关系包括numpy, scipy, matplotlib和tkinter, 这只是其中几个的名字. 随着Python语言的成熟, 现代工具开始失去与老版本Python的兼容性. 从16版开始, Amber的许多关键组件, 仅与Python 2.7,3.4(和更高版本)的版本兼容.
由于一些Linux版本附带的Python版本早于此版本, AmberTools包含一个configure_python脚本, 它将在Amber安装目录下载和安装一个兼容的Python版本, 以驱动Amber的Python组件. 如果标准配置过程没有检测到足够新的Python, 并且已经安装了足够的核心组件来运行大量的Amber Python功能, 则会询问您是否要配置下载并安装Miniconda(Continuum IO版).
如果您不是一位有经验的Python用户, 我们建议您选择"是", 并允许AmberTools创建和管理其所有工具所需的Python环境. 如果您是一位有经验的Python用户, 并且想要使用各种AmberTools Python API, 请参阅下面的将AmberTools Python包安装到标准Python环境的选项.
这是如果用户选择"是"AmberTools将安装的软件包列表: python2.7, numpy, scipy, cython, ipython, notebook, matplotlib. 总下载文件大小为〜100 MB, 总安装大小为〜0.5GB. 用户可以通过amber.python访问Python.
默认情况下, configure将尝试使用$AMBERHOME/miniconda/bin中的python可执行文件. 如果找不到, 则会通知并询问您是否要安装Miniconda. 上述完成后, AmberTools将会下载并安装这个版本的Python 2.7(并且将会在以后的配置调用中首先查找该版本, 除非被告知去别处查看).
您可以使用--with-python标志来配置(通过--with-python/path/to/python)以指定您希望用于Amber的特定Python可执行文件. 这个标志忽视了对现有先决条件的检查, 只有当你知道你在做什么时才应该使用它. 在这种情况下, 您可以自己安装所需的程序:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">#</span> Or via traditional"pip" package manager <span style="color:#A2F">#通过conda(推荐)</span> <span style="color:#A2F">conda</span> install <span style="color:#666">--file</span> AmberTools/src/python_requirement.txt <span style="color:#A2F">#或者通过传统的"pip"包处理</span> <span style="color:#A2F">pip</span> install <span style="color:#666">-r</span> AmberTools/src/python_requirement.txt </pre></div>默认情况下, AmberTools尝试将Python包安装到$AMBERHOME/lib/pythonX.Y, 其中X.Y是通过配置找到(或分配)的Python版本. 如果configure安装了一个Miniconda的版本, 就是$AMBERHOME/lib/python2.7. 然后将amber.sh和amber.csh资源脚本添加到您的PYTHONPATH环境变量中, 以确保Python运行时可以找到这些包. 在某些情况下, 像与标准的Phenix绑定, 外部程序可能依赖于Amber的Python软件包安装到该位置, 因此选择其他选项之一有可能会破坏第三方扩展. 对于有使用和开发Python经验的人来说, 这些问题很容易被发现和修复. 如果您不确定, 建议您保持默认行为.
然而, 在许多情况下, 用户可能会设置复杂的Python环境(例如通过conda或virtualenv), 并希望Amber的Python组件在它们的Python环境中更自然地配合使用, 而不是简单地转储到$AMBERHOME/lib/pythonX.Y. 对于这些情况, 已经添加了--python-install选项来进行配置, 该选项将用户, 本地或全局作为参数. 本地是默认行为. 有需要的用户通过命令安装所有的Python软件包.
这使得包可以Python运行时自动地被看到, 但是不需要修改任何可能需要root权限的系统级目录(不过, 它也不适用于任何其他用户帐户).
全局选项将会运行
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">python</span> setup.py install </pre></div>并尝试将所有Amber模块和程序包安装到根级Python站点包目录. 如果您使用的是系统Python(不推荐), 则这将需要root访问权限, 但如果使用本地安装的Python(如Anaconda或Miniconda), 则可能不需要任何特殊权限.
鼓励用户使用Python版本2.7和3.4(或更高版本), 因为这些版本已经验证可以与Amber的所有Python组件(假设满足其他先决条件, 如numpy和/或scipy)一起使用. 已知Python 2.6不能使用pytraj和ParmEd(以及依赖于ParmEd的任何包, 如MMPBSA.py).
AmberTools的不同组件支持的Python版本不同. 一些代码(如pytraj, ParmEd和pdb4amber)在Python 2.7和Python 3.x中均不变, 而其他代码在安装时需要使用2to3进行转换. 如果用户想将AmberTools(比如pysander, ParmEd)和Phenix, PyRosetta等第三方软件包结合起来, 用户需要使用Python 2.7版本, 因为这些软件包还不支持Python 3.x.
如果您选择"yes"允许配置从Miniconda安装Python, 或者您直接运行configure_python脚本, Python将安装到$AMBERHOME/miniconda. 在$AMBERHOME/bin中创建与主python, ipython, jupyter和conda可执行文件的符号链接, 分别命名为amber.python, amber.ipython, amber.jupyter和amber.conda. 这样做是为了避免与其他可能存在的Python环境发生潜在的冲突.
conda程序是用于安装新Python包的Python包管理器. 所以如果你需要安装一个新的软件包, 可以使用amber.conda程序来完成. 例如, 要安装pandas库, 请使用该命令
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">amber.conda</span> install pandas </pre></div>有关信息, 请参阅有关Anaconda(和Miniconda)的Continuum IO文档.
请参见https://www.continuum.io/documentation获得更多相关信息.
对于大多数用户来说, 只需运行配置脚本并对更新请求回应"是"就会自动下载并应用所有的修补程序. 本部分描述负责管理更新的主要更新脚本. 当您在邮件列表上寻求帮助时, 我们建议您至少要简略介绍基本用法的第一部分——特别是关于—version的标志.
AmberTools和Amber的更新是使用Python脚本update_amber自动下载, 应用和管理的. 该脚本适用于从Python 2.4到最新的Python 3版本的每个Python版本. $AMBERHOME中的配置脚本自动使用update_amber搜索AmberTools(当Amber存在时)可用更新, 除非使用--no-updates标志(必须是第一个配置选项)确定禁用. 当有任何可用的, 你会被询问你是否想要它们下载和应用. 这个脚本保留在$AMBERHOME中, 可以从任何地方执行(它将验证AMBERHOME设置是否正确), 但是如果从AMBERHOME移出, 它将不起作用. 有三种主要的操作模式或操作, 您可以使用它们执行:
$AMBERHOME/update_amber --check-updates: 此选项将查询Amber网站上发布的任何尚未应用于您的安装的更新. 如果你认为你已经发现了一个错误, 在发送问题之前, 这是有帮助的, 因为你的错误可能已经修复.
$AMBERHOME/update_amber --version: 此选项将返回到目前为止, 已经应用到当前树的修补程序. 在给Amber列表发送问题邮件时, 输出此命令非常重要, 因为这可以让我们确切知道已经应用了哪些更新.
$AMBERHOME/update_amber --update: 此选项将进入Amber网站, 下载所有尚未应用于您的安装的更新, 并将其应用于源代码. 注意, 您将不得不重新编译任何受影响的代码以使更改生效!
update_amber还具有附加功能, 可以更好地控制修补过程. 有关选项的完整列表, 请使用--full-help命令行选项. 这些被认为是高级选项.
$AMBERHOME/update_amber --download-patches: 只下载补丁, 不要应用补丁$AMBERHOME/update_amber --apply-patch = <PATCH>: 这将应用第三方补丁$AMBERHOME/update_amber --reverse-patch = <PATCH>: 逆转通过--apply-patch选项应用的第三方补丁文件(参见上文).$AMBERHOME/update_amber --show-applied-patches: 显示已应用的每个修补程序的详细信息(包括第三方修补程序)$AMBERHOME/update_amber --show-unapplied-patches: 显示已下载但尚未应用的每个修补程序的详细信息.$AMBERHOME/update_amber --remove-unapplied: 删除所有已下载但未应用的修补程序. 这将强迫update_amber下载该补丁的新副本.$AMBERHOME/update_amber --update-to AmberTools /#, Amber /#: 此命令将AmberTools升级到特定版本所需的所有修补程序, 并将Amber升级到特定版本. 注意, 没有更新将会逆转此命令. 您只能指定AmberTools版本或Amber版本(或两者, 逗号分隔). 没有补丁被应用到一个被忽略的分支.$AMBERHOME/update_amber --revert-to AmberTools /#, Amber /#: 这个命令与上面描述的-Update相同, 除了它只会你逆转补丁, 不会应用它们.update_amber还会根据详细设置提供有关每个修补程序的不同数量的信息. 详细级别可以使用--verbose标志设置, 可以是0到4之间的任何整数. 默认的详细级别根据必须描述的更新数量而变化. 如果只需要描述少量的更新, 所有的细节都会被打印出来. 必须描述的更新越多, 打印的信息就越少. 如果您在命令行上手动设置一个值, 它将覆盖默认值. 这些值如下所述(每个级别在添加附加信息之前打印所有级别的信息):
0: 仅打印更新文件的名称(没有其他信息)1: 还打印出受影响程序的名称2: 还打印出该更新作者所写更新的描述.3: 同时打印创建修补程序的人员名称和创建日期.4: 还打印出由修补程序修改的每个文件的名称.update_amber需要连接到互联网, 它会检查是否可以在10秒内联系http://ambermd.org. 如果不是, 则会报告错误并退出. 如果您的连接速度特别慢, 您可以通过--timeout命令行标志(时间以秒为单位)来延长此超时时间.
Proxies默认情况下, update_amber将尝试通过与wget和curl程序相同的机制来联系互联网. 对于通过代理服务器连接到互联网的用户, 您可以自己设置http_proxy环境变量(在这种情况下, 您可以在此处忽略有关代理的其他建议), 也可以配置update_amber为通过代理连接到互联网. 要设置update_amber以通过代理连接到Internet, 请使用以下命令:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">$AMBERHOME/update_amber</span> <span style="color:#666">--proxy</span> = <PROXY_ADDRESS> </pre></div>您可以经常从您的IT部分或者用于浏览网页的您最喜爱的(已配置)网络浏览器中的首选项找到您的代理地址. 如果您的代理已通过身份验证, 则还需要设置一个用户:
<div class="highlight"><pre style="line-height:125%"><span style="color:#A2F">$AMBERHOME/update_amber</span> --proxy-user = <USERNAME> </pre></div>如果您已经设置了用户名来连接到您的代理, 那么update_amber首次尝试使用在线资源时, 系统会要求您输入代理密码. (为了安全起见, 您的密码永远不会被存储, 并且每次update_amber运行时都需要重新输入).
您可以使用--delete-proxy命令行标记清除所有代理信息——实际上只有在您不再需要通过任何代理进行连接时才是必需的, 因为每次配置特定的代理用户或服务器时, 都会覆盖之前所设置的任何代理.
Mirrors如果要从其他网站下载Amber修补程序, 或者甚至是从本地文件系统下载文件夹, 则可以使用--amber-updates和--ambertools-updates命令行标志来指定特定的Web地址(必须以http : //)或本地文件夹(使用绝对路径). 您可以使用--reset-remotes命令行标志擦除这些设置, 并返回到http://ambermd.org上的默认Amber位置.
如果您设置了在线镜像, 并且从未计划直接连接到http://ambermd.org, 当它使用--internet-check命令行选项验证Internet连接时, 你可以更改update_amber尝试连接到的网址.
2.4 联系开发人员
请将建议和问题发送至amber@ambermd.org. 你需要注册才能在那里发帖; 注册, 请访问http://lists.ambermd.org/mailman/listinfo/amber. 你可以在同一个网站上退订这个邮件列表.