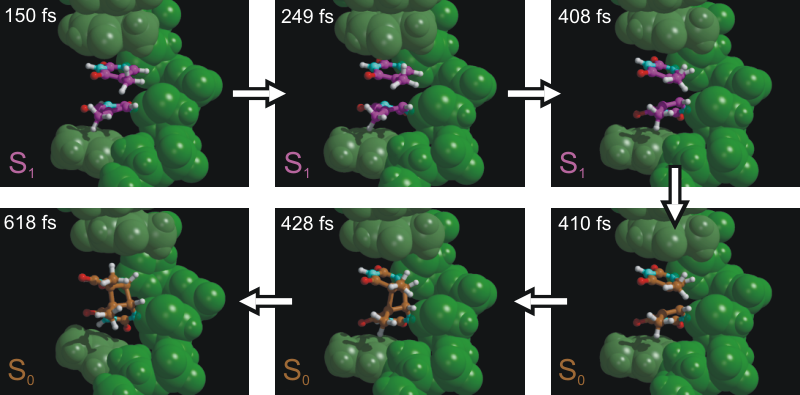
链内胸腺嘧啶二聚体化(图1)被认为是在紫外辐射下导致DNA损伤的最常见的过程. 胸腺嘧啶二聚体的形成具有潜在的重要生理学意义. 这个诱变的光产物能干扰DNA发挥功能, 从而引发复杂的生物学响应, 包括凋亡、免疫抑制和致癌作用.
图1. 吸收紫外光导致胸腺嘧啶形成二聚体, 快照来自从头算分子动力学模拟
为了暴露于UV辐射下仍能存活, 生命体已经进化出了修复DNA损伤的复杂机制. 初始步骤通常是探测到一个损伤点, 如胸腺嘧啶二聚体. 接下来, 这个二聚体或者被修复, 或者被完全清除.
光解酶(Photolyas)能够通过与其结合来探测二聚体位点, 然后对二聚体剪接过程进行催化, 使其恢复为原始的嘧啶碱基(图2). 光解酶含有还原的黄素辅酶辅因子(reduced flavin co-enzyme co-factor), 当吸收UV光时, 可以向结合的胸腺嘧啶二聚体提供电子. 恢复为原始的胸腺嘧啶碱基后, 电子会流回黄素, 光解酶也可以继续修复下一个病变.
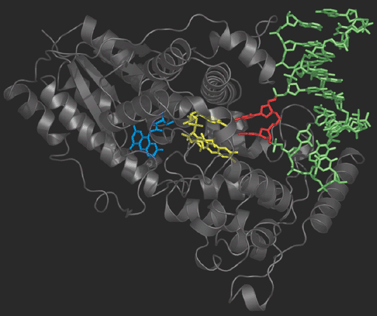
图2. 光解酶-DNA复合物. 光解酶使用紫外光催化二聚体恢复为原始胸腺嘧啶碱基(红色)的过程.
在没有光解酶的帮助下, 胸腺嘧啶二聚体也可以恢复. 在这种所谓的自我修复过程中, 二聚体摄入电子时自发分裂. 依赖于碱基序列, 这种电子容易获得. 在本教程中, 我们将通过从头算分子动力学模拟来考察这种自我修复过程.
本教程旨在学习用于研究生物体系的基本QM/MM技能, 共包括八个部分:
本教程中我们将使用如下软件包GROMACS, rasmol, pymol, B.
B(原名Biomer)是一种基于Java的在线生物分子建模程序包, 可用于生成生物聚合物和有机小分子的初始结构. 我们将使用B来生成双链DNA寡聚体. 你可以自由地选择任意的DNA序列, 只要其中一条链上至少有一个TT重复单元即可. 我们在本教程中提供的示例文件基于A20T20低聚物.
请参阅B手册和常见问题页面了解模型构建方法. 将你的DNA分子保存为pdb格式. A20T20低聚物的pdb文件可以在这里下载.
我们使用Amber99力场描述原子间的相互作用. GROMACS的Amber接口可以从Eric Sorin的网站下载.
为了使pdb2gmx工具能够识别胸腺嘧啶二聚体, 我们需要修饰ffamber99.rtp残基数据库. 我们引入两个新的胸腺嘧啶残基dtA和dtB, 并在C5原子和C6原子之间定义额外的碱基间键. 修改后的ffamber99.rtp可在这里下载.
使用下面的pdb2gmx命令, 我们创建拓扑文件topol.top和构型构型文件conf.gro
pdb2gmx -ignh -f Bmodel.pdb
其中的-ignh选项忽略输入文件中的氢原子, 并强制pdb2gmx基于amber99.hdb数据库添加氢原子.
我们首先进行真空中的能量最小化, 使用最速下降算法. 查看我们所用的steep.md文件, 熟悉我们执行的最小化过程.
先执行grompp, 然后mdrun进行能量最小化
grompp -f steep.mdp
mdrun -v -c minimized.pdb
能量最小化后的DNA结构可在这里下载.
下一步是添加水分子并保证体系电中性. 我们使用GROMACS工具editconf, genbox和genion.
首先将能量最小化后的结构置于方形盒子的中心
editconf -c -d 1.6 -f minimized.pdb -o boxed.pdb
然后, 添加水分子, 使用tip4p模型
genbox -cs tip4p.gro -cp boxed.pdb -p topol -o solved.pdb
最后, 需要加入38个钠离子来中和体系的负电荷. 先使用neutralize.mdp创建一个tpr文件
grompp -f neutralize.mdp -c solved.pdb -p topol
得到topol.tpr文件后, 可以使用genion将38个抗衡离子随机放在盒子中
genion -np 38 -pname Na -o neutral.pdb -random
我们必须修改拓扑文件, 手动添加38个钠离子(NA), 并去除38个tip4p水分子. 最终的拓扑文件可以在这里下载. 接下来, 我们还需要一个索引文件, 其中包含DNA, 水和离子的原子编号.
由于水分子和DNA原子之间可能存在重叠, 我们必须对体系进行能量最小化. 我们再次使用最速下降法来消除初始结构中可能的应变. mdp文件可以在这里下载.
grompp -f mini_sol.mdp -p neutral.top -c neutral.pdb -n neutral.ndx
mdrun -v -c neutral_mini.pdb
然后, 我们使用equi_sol.mdp中的参数对水和离子进行100 ps的平衡. 在溶剂平衡期间DNA中的重原子会被位置限制.
grompp -f equi_sol.mdp -p neutral.top -c neutral_mini.pdb -n neutral.ndx
mdrun -v -c neutral_sol_equi.pdb
A20T20低聚物溶剂平衡后的结果可在这里下载.
我们将对体系进行1 ns的经典分子动力学模拟, 其中包含由20个碱基对组成的带胸腺嘧啶二聚体的DNA分子, 水和离子. 作为模拟的起始结构, 我们使用上一步骤的平衡结构.
grompp -f md.mdp -p neutral.top -c neutral_sol_equi.pdb -n neutral.ndx
mdrun -c equi.gro
注意, 我们现在使用gro文件格式, 因为我们在下一步中不仅要使用平衡后的坐标, 还要使用相应的速度.
我们现在将设置用于GROMACS QM/MM模拟的体系. 胸腺嘧啶碱基二聚体使用半经验AM1理论水平进行描述, 而系统的其余部分使用Amber99力场. 图3显示了我们如何将体系划分为QM和MM部分.
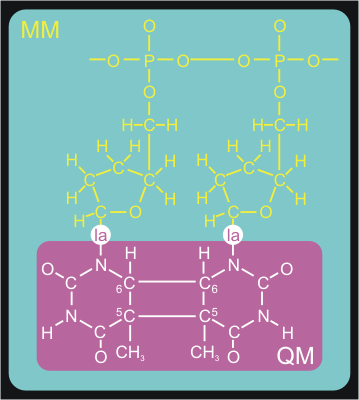
图3. 将整个体系划分为QM子体系和MM子体系. 使用QM半经验水平描述QM子系统, 而由反应物脂肪族尾部组成的其余体系部分使用Amber99力场建模. 在QM/MM边界处引入链接原子对QM子体系进行封端.
QM/MM划分破坏了体系中的化学键. 因此, 我们需要用所谓的链接原子(图3中的la)对QM子体系进行封端. 链接原子在QM计算步骤中被视为氢原子, 并不真实存在于MM子体系中. QM步骤中计算得到的链接原子上的力, 会被重新分配到所在键的两个原子上. 键长自身在计算过程中受到约束.
要使用GROMACS的QM/MM功能, 我们必须
在连接QM和MM子体系的化学键处, 我们引入了链接原子. 在GROMACS中, 我们将链接原子视为一个特殊的原子类型, LA. 该原子类型在QM计算中被视为氢原子, 而在力场计算中被视为虚原子. 链接原子(如果存在)是体系的一部分, 但与体系中的任何其他原子没有相互作用, 除了作用在其上的QM力会重新分配到所在键的两个原子上. 因此, 在拓扑中链接原子(LA)被定义为虚原子:
[dummies 2]
LA QMatom MMatom 1 0.73
注意, 链接原子没有质量.
此外, MM和QM原子之间的键仍维持在力场水平:
[ bonds ]
QMatom MMatom 1
注意, 因为在我们的体系中, QM/MM键是碳-氮键(0.153 nm), 我们使用的约束长度为0.153 nm, 虚拟位置为0.65. 后者是理想C-H键长和理想C-C键长之间的比. 使用这一比例, 链接原子离QM原子对距离始终为0.1 nm, 与碳-氢键长度一致. 如果QM和MM子体系通过其他类型的键相连接, 则需要使用与其键类型匹配的约束和虚拟位置. 此外, 由于在模拟的每一步中都会重新计算链接原子的位置, 因此在初始构型文件中链接原子的位置并不重要. 我们可以简单地将两个链接原子置于原点(0,0,0).
最后, gromac的自带的amber力场文件中不包括链接原子. 因此, 我们需要手工添加. 在ffamber99.atp文件的末尾添加
LA 0.0 ; Link Atom for QM/MM
这样如果输入结构中存在链接原子, pdb2gmx就能够识别了. 我们还要将链接原子添加到ffamber99nb.atp文件
LA LA 1 0.0000 0.0000 A 0.0 0.0
这样grompp就能够知道对链接原子使用的Lennard-Jones参数. 很明显这些参数应该为零. 第一个整数是QM/MM计算中使用的元素类型, 链接原子被视为氢(1).
下载需要的AMBER力场文件amber.tar. 下载后使用命令tar -xvf amber.tar解压.
一旦我们决定了哪些原子应该使用QM方法进行处理, 我们就将这些原子, 包括链接原子(如果有的话), 添加到索引文件. 我们可以使用make_ndx程序, 或者手动将原子编号加入index.ndx文件中. 下载本教程中使用的索引文件qmmm.ndx. 可以单独约束QM子体系中的键, 也可以不限制而同时限制MM子体系中的键. 如果QM原子可能会经历键的断裂/形成反应, 不限制QM子体系中的键非常必要. 在这种情况下, GROMACS的键类型5用于QM子体系中的键:
[ bonds ]
QMatom1 QMatom2 5
QMatom2 QMatom3 5
设置GROMACS执行QM/MM计算的最后一步是, 指定GROMACS使用什么级别的QM理论处理QM子体系, 使用哪种QM/MM接口, QM子体系的多重度, 等等. 所有这些都在mdp文件中定义. 运行QM/MM时需要包括以下选项:
QMMM = yes
QMMM-grps = QMatoms
QMmethod = RHF
QMbasis = STO-3G
QMMMscheme = ONIOM
QMcharge = 0
QMmult = 1
请注意, 上面给出的是默认选项, 实际所用选项取决于你的体系. 你可以参考我们用于短时间QM/MM平衡模拟的mdp文件qmmm1.mdp. 如果选择半经验的QM方法, 如AM1或PM3, 应忽略QMbasis.
使用上面的设置, 我们将进行1 ps的短时间QM/MM MD模拟.
我们现在对体系进行1 ps的短时间QM/MM(AM1/Amber99)平衡模拟.
为此, 我们需要一个已链接mopac库的特殊的mdrun二进制文件.
要使下载的mdrun变为可执行文件, 请使用
chmod +x mdrun
我们仍然使用正常版本的grompp
grompp -f qmmm1.mdp -p qmmm.top -n qmmm.ndx -c qmmm1.gro
./mdrun -v -c qmmm1out.gro -x traj_stepIV.xtc
下载qmmm1.mdp, qmmm.top, qmmm1out.gro
由于此模拟大约需要40分钟才能完成, 我们给出打包好的输出文件供你参考. 你既可以使用这些文件进行下一步骤(先解压tar -xvf qmmmmd.tar), 也可以等待自己的模拟结束, 使用自己得到的文件进行下一步.
在下一步中, 我们将使用1 ps平衡后的结构来研究过量电子对胸腺嘧啶二聚体的稳定性的影响.
电子转移到胸腺嘧啶二聚体之后, 之前中性QM子体系的总电荷降为-1. 此外, 过量电子将使QM子体系的总自旋量子数从1/2增加到1. 因此, 现在体系的多重度为双线态而不是单线态. 这些改变必须添加到mdp文件的QM/MM参数中
QMMM = yes
QMMM-grps = QMatoms
QMmethod = AM1
QMbasis = STO-3G
QMMMscheme = ONIOM
QMcharge = -1
QMmult = 2
我们现在对胸腺嘧啶二聚体上带有过量电子的体系进行1 ps的短时间QM/MM模拟. 我们使用之前QM/MM模拟的最后一帧作为起始结构:
grompp -f qmmm2.mdp -p qmmm.top -n qmmm.ndx -c qmmm1out.gro
mdrun -v -c qmmm2out.gro -x traj_stepV.xtc
此模拟仍然需要大约40分钟时间, 因此你可以下载我们得到的输出文件electron1.tar, 并使用前面的方法解压.
在最后一帧中, 我们看到环丁烷环中的一根键断裂了(图4). 为了观看整个过程, 你可以将轨迹转换为pdb格式, 然后使用pymol或VMD载入.
trjconv -s -f traj_stepV.xtc -n qmmm.ndx -o traj2.pdb
提示时选择heavyatoms组.
因此, 摄入电子后, 二聚体上的C5原子之间的键(图2)断裂. 考虑到断键发生的时间尺度, 该过程肯定是无垒的. C5-C5键而不是C6-C6键断裂的原因可能是甲基之间的空间排斥. 在模拟的时间尺度上, C6原子之间的共价键仍保持完整. 因此, 断裂第二根键是涉及(小)活化势垒的活化过程. 为了在短时间的QM/MM MD模拟中克服这个能垒, 我们将使用化学洪泛方法, 这将在下一节中描述.
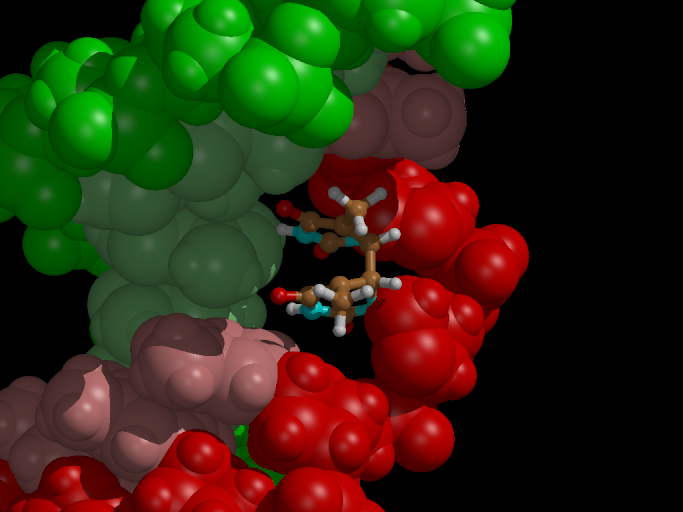
图4. 摄入过量电子会导致胸腺嘧啶二聚体之间的环丁烷环中的一根键超快断裂.
QM/MM MD模拟的主要瓶颈在于, 由于涉及巨大的计算量, 只有在皮秒到纳秒或更快时间尺度内发生的过程可以直接研究. 不幸的是, 除少数情况外, 其他相关过程, 如化学反应发生在更慢的时间尺度, 因此目前远远不能使用传统的QM/MM MD进行研究.
化学洪泛技术通过在QM/MM势能表面上增加洪泛势来解决这个问题. 这种洪泛势局部地破坏离解状态, 从而能显著地加速体系从初始能量最小点的逃逸而不影响反应途径. 洪泛方法的一个显著优点是不涉及过渡态的先验知识, 尤其是不必假定或猜测反应坐标、过渡态、中间体或产物状态. 因此, 该方法可以无偏差地预测过渡途径和产物状态.
洪泛涉及两个步骤:第一, 对体系的自由能形貌使用准简谐近似. 第二, 根据该近似构建多变量的洪泛势 $V_{fl}$, 并用它提升离解势阱的底部, 而不影响更高能量的区域, 特别是围绕势阱周围, 决定过渡态途径的势垒.
对于自由能面的准简谐近似, 选择线性集约坐标. 这些坐标可通过主成分分析(PCA)获得. 选择了合适的集约坐标后, 会构造高斯形的洪泛势, 使得其主轴平行于集约坐标.
我们首先需要一组集约坐标来表征QM子体系中的相关运动. g_covar程序可以从MD轨迹计算涨落运动的协方差矩阵. 由于显而易见的原因, 我们将分析限制在QM区域. 此外, 我们仅考虑重原子的运动. 对角化协方差矩阵, 得到的特征向量就可以作为集约坐标.
g_covar -n qmmm.ndx -f traj_stepV.xtc -s
提示时选择重原子组进行叠合和分析.
特征向量(或集约运动)可以使用pymol可视化. 为此, 我们使用g_anaeig创建这些运动的一段小轨迹. 例如命令
g_anaeig -nframes 40 -first 1 -last 1 -n qmmm.ndx -extr vector1.pdb
提示时选择heavyatoms组. 运行完成后. 会为QM子体系的第一个集约自由度生成内插的轨迹.
我们将使用所有的集约坐标或特征向量来构造洪泛势, 其强度为150 kJ/mol. make_edi程序可生成带有洪泛参数的sam.edi文件.
make_edi -f eigenvec.trr -eig eigenval.xvg -flood 1-48 -Eflnull 150 -outfrq 1 -s -n qmmm.ndx -tau 0
-outfrq选项设置输出记录的频率, 我们希望在模拟的每一步都输出. 生成的sam.edi文件可由mdrun读入并进行洪泛模拟, 我们将在下面看到.
我们对所有集约自由度应用洪泛势并继续QM/MM模拟. 我们使用先前QM/MM模拟的最后一帧作为洪泛模拟的起始结构:
grompp -f qmmm2.mdp -p qmmm.top -n qmmm.ndx -c qmmm2out.gro
./mdrun -v -c qmmm3out.gro -ei sam.edi -x traj_VII.xtc
下载qmmm2.mdp, qmmm2out.gro, qmmm3out.gro, sam.edi
这一模拟仍然需要大约40分钟的时间, 你可以下载我们得到的输出文件electron2.tar.
图5为洪泛模拟的最后一帧. 我们可以看到碱基之间的两根共价键都断开了. 然而, 其中一个碱基上仍然存在过量电子, 这通过嘧啶氮原子的sp3杂化反映出来.
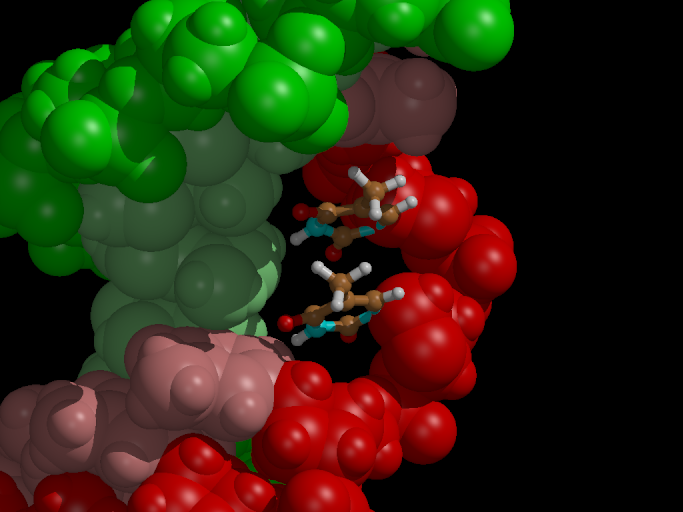
图5. 利用洪泛势, 胸腺嘧啶二聚体在2 ps内被完全破坏.
实际上, 电子会被转移回光解酶中的FADH辅因子, 或DNA链中的相邻碱基. 为了在我们的模拟中考虑这种反向传递, 我们从最后一帧继续模拟, 使用旧的mdp文件, 其中的QM子体系为中性单线态.
grompp -f qmmm1.mdp -p qmmm.top -n qmmm.ndx -c qmmm3out.gro
./mdrun -v -c qmmm4out.gro
下载qmmm4out.gro, 输出文件final.tar
模拟的最后一帧如图6所示. 我们看到DNA完全恢复了!
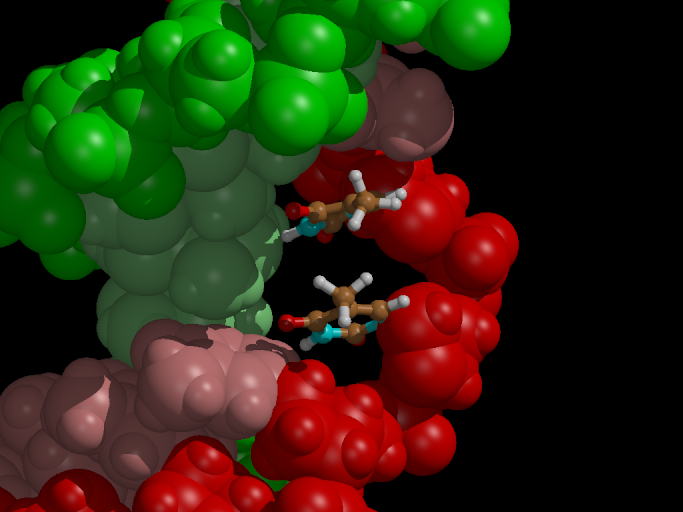
图6. 恢复的DNA.
我们对含胸腺嘧啶二聚体病变的DNA进行了QM/MM MD模拟. 我们发现, 二聚体摄入电子会导致维持二聚体结构的两根键之一快速断裂. 第二根键的断裂存在势垒. 为了加速跨越势垒, 我们使用洪泛技术. 根据洪泛势的强度以及克服势垒所花的时间, 可以估计出势垒高度的上限. 如果你对此感兴趣, 我们建议你阅读Helmut Grubmueller的论文, 是他发展了洪泛技术.
根据洪泛模拟, 我们可以推断反应机理. 因此, 洪泛轨迹为更高等的方法, 如伞形采样或过渡途径采样提供了良好的起点.
我们希望你享受学习这个教程的过程, 并发现它对你现在或未将来的工作有所帮助. 如果在进行QM/MM计算时遇到麻烦, 或想讨论一些问题, 你可以随时与我联系.