toc {:toc}
原始文档: Molecular Modeling Practical 多肽-蛋白; Molecular Modeling Practical 泛素耦合酶
本教程用于引导学生运行蛋白质的分子动力学(MD)模拟. 如果待研究蛋白不包含非标准氨基酸残基, 教程所用的流程可作为研究蛋白的一个良好起点. 完成本教程后, 学生应该能够知晓设置和运行模拟所涉及的步骤, 以及在不同模拟阶段应如何选择. 此外, 学生还应该知道如何对模拟结果进行核查以保证模拟质量, 并了解可以使用哪些分析方法, 哪些分析方法更适合自己的需要.
本教程的目的在于考察一段小肽结合到其蛋白受体时, 生物分子相互识别的两种机理--构象选择以及诱导匹配--所具有的不同贡献. 完成本教程后, 学生应该能够:
大多数教程都是使用终端或控制台应用程序完成的, 因为我们用于准备和进行模拟的程序没有图形化界面. 为了便于了解这种新的运行环境, 我们准备了一份简单的帮助文档, 以便让你熟悉在Linux环境下"生存"所需的基本命令. 点击这里下载此文件.
Linux下的所有命令都需要小心输入, 因为shell(负责解析命令的程序)是区分大小写的. 另一经常发生的错误是将数字0当成了大写字母O, 小写字母l当成了数字1, 或者反过来. 你可以简单地复制粘贴这些命令: 先使用鼠标选中它们, 然后在需要输入的地方按下鼠标中键(Linux模式, Windows下未必可行). 首先来试试下面的命令:
这会列出你的当前用户名. 请确保你不是以root用户登录.
上面的命令列出当前工作目录下的所有文件和文件夹. 当遇到file not found之类的错误时, 可以使用这个命令来检查.
这个命令给出当前目录的完整路径.
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">cd</span> Desktop </pre></div>此命令展示了如何更换目录.
在决定细胞命运的大量蛋白-蛋白相互作用中, 多肽起着关键作用, 并占了大约40%的比例. 从辅激活剂到抑制剂, 它们在许多信号和识别途径中都有涉及, 并且已经被证实与大量蛋白结构域存在相互作用, 例如, MHC, SH3和PDZ结构域就是由于其多肽亲和能力而知名. 由于功能的多样性以及间接参与的许多生物途径的重要性, 使得多它们与许多疾病相联系. 药物设计中的一个领域就专门研究多肽并用于治疗各种疾病. 与小分子抑制剂相比, 多肽的优点在于可以模拟蛋白结合的结构域, 并且自身足够大能竞争性地抑制蛋白蛋白相互作用. 许多药物先导分子就包含抗菌多肽.
尽管科学家们收集了蛋白多肽相互作用的大量数据, 确定它们形成的复合物的结构仍然是个挑战. 这主要源于两个障碍, 多肽柔性极高, 并且与其底物的相互作用很弱, 这突出了它们在信号传导或调节中的重要性, 因为这些功能通常依赖于瞬态过程. 这些障碍使得实验结构测定并不简单, 需要借助额外的计算方法如生物分子对接才能完成. 从建模的角度看, 常规的用于蛋白-配体或者蛋白-蛋白对接的算法也经常受到柔性问题的困扰.
解决柔性问题的一个途径是进行分子动力学模拟, 对多肽的构象空间进行采样, 收集具有代表性的构象, 并用于预测它们与蛋白受体的相互作用.
上个世纪人们提出了一些理论来解释分子的识别过程. 在这些理论中, 构象选择 和 诱导匹配 机理在过去的50年中得到了大致相同多的支持. 构象选择假定, 在配体不存在的情况下, 蛋白处于多种离散构象状态的平衡中, 其中包括倾向与配体结合的那个构象. 这一概念与诱导匹配理论相反, 后一理论起初引入用于描述酶的活性, 认为构象匹配是由与底物的结合导致的.
在本教程中, 我们要考察这一两难问题--诱导匹配和/或构象选择, 方法是使用不同初始构象的分子动力学模拟对多肽的构象空间进行采样. 我们将不同的轨迹与实验确定的多肽在与其蛋白受体形成的复合物中的构象(结合构象)进行比较, 并检查这一构象在MD过程中是否出现, 这样就能验证构象选择理论.
经典分子动力学模拟从定义好的构型开始, 使用牛顿运动方程来计算粒子的轨迹. 对于体系中的每一粒子, 所受的合力根据它与其他粒子的相互作用来计算, 这些相互作用以力场进行描述. 力除以粒子的质量为加速度, 再加上粒子以前的位置和速度, 就决定了一小段时间步长之后粒子的新位置. 高的时间和空间分辨率使得MD模拟在测试基于实验数据的模型, 理解功能背后的原理并形成新的假说时非常有用. 不幸的是, 模拟体系的大小以及时间尺度存在限制.
本教程使用GROMACS进行MD模拟和分析, 它是一个可以免费获取程序包, 遵循GNU GPL(General Public License). GROMACS程序只有命令行界面, 这意味着每一步运行都要键入程序的名称及其选项. 注意命令是区分大小写的, 每一命令键入时都必须精确地与教程一致, 关于GROMACS的更多信息以及手册请见其官方网站. (提示: MPI并行版本的GROMACS可以在多个节点(集群中的机器)上分配任务从而提高性能, 对单机而言MPI并不能提高性能, 因为GROMACS可以自动使用单个机器的多个核心)
由于程序只有命令行界面, 因此我们必须使用终端. 尽管在Windows环境下借助DOS终端也可以运行GROMACS, 使用Linux有一些优点, 这也是本教程的选择. 对一些学生来说, 从Windows转到Linux或许会感到别扭, 因为他们已经更习惯了Windows提供的界面. 重要的一点是记住, Linux并非Windows的免费克隆品, 而是一个功能强大, 高度可定制化的操作系统, 能够使你的计算机发挥更大的威力. 从Windowa转到Linux有时会被描述成摩托车换汽车. 开始使用Linux终端时, 需要知道一些最基本的命令(ls, cd, mkdir, cp, mv, rm, more). 更多信息请查看这里和这里, 也可以参考上面提供的帮助文档.
默认情况下, 你的系统已经正确设置好了.
你可以使用gmx luck命令测试GROMACS是否正确安装. 幸运的话, 你会得到一句引言. 这说明你的GROMACS已经正确安装好了.
在进行其他操作之前, 必须要获得初始结构. 结合构象可以从蛋白数据库获取, 它是一个蛋白三维结构的数据仓库, 根据每个蛋白的登记号(如1klu)搜索蛋白(提示: 你可以使用命令下载文件: wget http://www.rcsb.org/pdb/files/1klu.pdb).
蛋白数据库的页面, 除了提供结构的坐标文件(pdb文件)外, 也包含了分子体系的一些有用信息, 如所有分子的一级序列, 二级结构的指认, 长度, 生物功能等. 花些时间来浏览页面, 收集待研究分子的尽可能多的信息.
为开始教程, 根据多肽的PDB号下载相应的结构, 并记下下列信息:
首先使用分子查看软件来看看分子的结构, 我们建议使用PyMOL. 可使用下面的PyMOL命令来载入结构
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">PyMOL</span> 1klu.pdb </pre></div>执行上面的命令后, PyMOL会启动, 在一个窗口中使用线形模式来显示分子, 并在主窗口的右面列出各个对象, 这些对象可以通过点击其名称来移除. 每个对象名称的后面是菜单, 用于更改显示模式. 试着使用卡通模式来显示结构. 对那些习惯使用键盘的人(我们强烈建议你也使用这种方式), 可以通过在窗口中键入下面的命令来达到目的
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">show</span> cartoon </pre></div>使用上一步骤中收集到的信息, 通过键入下面的命令来选择包含多肽的pdb链
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">select</span> sel_peptide, chain X </pre></div>然后抽取多肽链保存到另一个对象
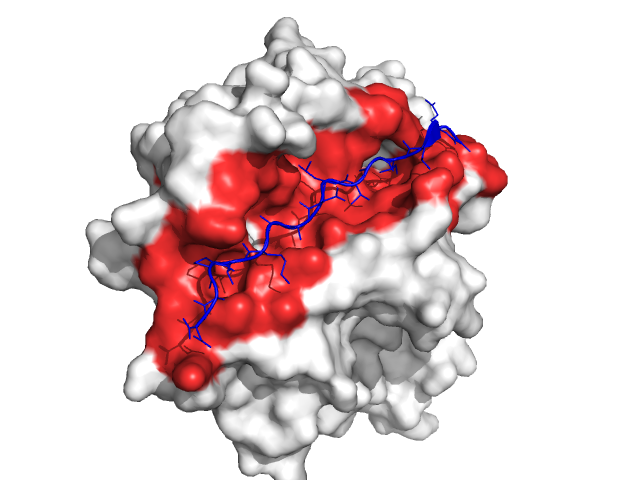
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">extract</span> peptide, sel_peptide </pre></div>计算并显示蛋白的溶剂可及表面
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">as</span> surface, 1klu </pre></div>对蛋白表面和多肽使用两种可区分的颜色进行着色
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">color</span> white, 1klu <span style="color:#AA22FF">color</span> tv_blue, peptide </pre></div>选择处于多肽5埃之内的所有蛋白残基进行着色, 这构成了蛋白-多肽的界面
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">select</span> interface, byres( 1klu within 5A of peptide ) <span style="color:#AA22FF">color</span> red, interface </pre></div>如果对显示结果满意, 你可以进一步使用ray命令改善显示结果, 并使用png filename命令保存图片.
使用下面的命令记下在PyMOL中显示的结合多肽的氨基酸序列
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">set</span> seq_view, on </pre></div>比较PyMOL显示的序列和蛋白数据库给出的序列, 二者是否相同? 为什么?
PyMOL也可以用于生成多肽的初始构象. 默认情况下, 你可以使用Build菜单, 选择构象和需要增加的残基. 此外, 我们提供了一个脚本build_seq, 可以对给定的氨基酸序列生成相应的理想构象. 这个脚本可能很有用, 因为对于一些多肽构象, 如聚脯氨酸II, PyMOL并不知道所需要的phi/psi角度组合. 你可以通过在窗口中键入下面的命令来获知如何使用这个脚本
使用这个脚本生成多肽的三个不同构象, 我们在教程中要使用它们, 所以要给它们指定不同的名字, 将下面命令中的XXXXXX替换为结合多肽的序列(PyMOL中有显示)
使用卡通模式显示新创建的多肽
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">show</span> cartoon, peptide_* </pre></div>使用align命令在空间中对齐并叠合不同的构象, 查看它们的差异程度有多大
对齐与叠合计算会返回原子位置的根均方偏差(RMSD), 它定量地表征了不同构象的结构异同. 请记下每个align命令给出的RMSD值.
最后, 使用save命令保存构象, 类似下面保存螺旋多肽的命令
得到的pdb文件可以用于开始准备模拟.
现在使用命令quit退出PyMOL. 正如你注意到的, 绘制结构必需的所有信息都保存在.pdb文件中. 你可以使用gedit来查看pdb文件, 并试着理解这种文件格式.
pdb文件包含了很多涉及蛋白, 用于确定结构的实验方法和条件等的信息. PDB数据库网页实际就是将这些信息展示给你的. pdb文件中也包含了每一个原子的直角坐标. 注意, pdb文件中一般不包含成键连接信息, 而PyMOL像大多数分子查看软件一样绘制了原子之间的成键, 这些成键是自动根据原子间的距离来确定的.
结合态多肽的pdb文件和你自己使用
build_seq生成的有什么不同(如氢原子)?
为你的结构创建一个目录. 由于最后需要将结果合并起来, 最好通过组合PDB标识(如peptide_bound, peptide_helix等)和个人标识指定明确且唯一的目录名称. 将结构文件复制到目录中并切换目录. 举例来说,
现在开始真正的MD部分. 记住在每一步都要指定正确的文件名. 特别注意, 教程简单地使用protein.pdb和protein-EM-solvated.gro作为通用名称, 这些通用名称应该替换为待研究蛋白的名称(如bound_peptide.pdb和bound_peptide-EM-solvated.gro).
请仔细阅读教程, 并检查在每一步是否成功完成. 请细心阅读程序输出!!! 如果程序给出错误信息, 这些信息通常是自明, 容易理解. 检查文件类型和程序输出以理解每一步的过程. 大多数文件是可读的, 除了扩展名为.tpr, .xtc, .trr和.edr的文件
分子动力学(MD)模拟包含三个步骤: 首先, 必须准备好输入数据(模拟体系); 其次, 必须对准备好的体系运行成品模拟; 最后, 必须分析模拟结果并将其置于相关背景中. 尽管第二步是计算中最耗时的部分, 一些模拟常需要运行几个月, 实际上最费劲的步骤在于模拟体系的准备和模拟结果的分析. 本教程的这一部分提供准备蛋白MD模拟, 运行, 分析结果的一个示例.
本教程使用GROMACS 5.1.4版本, 大多数命令来自这一程序. 所采用的工作流程更通用, 也可以用于其他MD程序. 每一步骤会使用一个有输入和输出的节点示意地表示出来. 一个步骤可以是执行单个程序, 也可以是几个程序的组合, 其中一个的输出作为另一个的输入. 在单个程序的情况下, 会指定输入/输出类型和控制选项. 对大多数程序/步骤, 只会列出使用的输入/输出选项. 大多数程序提供了更多高级控制选项, 这里不再细述. 如果你有兴趣, 可以参看每个程序的帮助页面, 里面给出了程序功能的完整列表和说明. 这些帮助页面可以使用程序的-h选项来显示. 在工作流程中, 程序以黄色背景上的名称来标识, 输入/输出数据以绿色背景上的描述性名称标识, 蓝色块表示输入数据, 基本不依赖于待研究的结构. 橙色块是包含多个步骤及相关程序的过程. 在这种表示方法中, 橙色块单独用于表示模拟步骤, 在下面会详细解释. 数据到过程的连接线, 每一程序输入或输出的命令行选项使用黑色背景上的白色标识.
准备模拟体系是MD中最重要的步骤. 执行MD模拟可以在原子尺度洞察动力学的过程, 用于理解实验观察到的现象, 验证理论假说, 或者为一个有待实验验证的新假说提供基础. 然而, 对于上述各种情形, 设计的模拟必须适合目的, 要根据实际情况对模拟过程进行设计, 这意味着设置模拟的时候必须十分小心.
缺失残基/侧链/原子, 非标准基团
本教程我们模拟的是多肽. 整个过程的首要步骤是选择初始结构, 如前面所说. 然后就要检查这个结构是否缺失残基和原子, 这些残基和原子模拟时必须考虑, 因此必须想办法补充完整. 从pdb库下载的文件中会列出缺失的残基和原子. 由于本教程不涉及蛋白建模, 因此所用的初始结构是完整的, 不存在缺失, 因此无需进一步处理.
另一个需要注意的问题是, pdb结构文件中可能包含非标准残基, 修饰过的残基, 溶剂分子或者配体. 这些基团没有力场参数可用. 如果存在这样的基团, 要么除去它们, 要么补充它们的力场参数, 这牵涉到MD的高级课题. 本教程假定结构不含这类基团, 只包含天然氨基酸.
最后, 一些晶体散射数据的分辨率足够高, 能够在密度网格中区分水分子. 通常这些水分子只以氧原子表示, 在开始准备步骤前必须去除它们, 除非出于特殊目的要保留它们. 对本教程的多肽不需要考虑这种情况.
幸运的是, 大多数的这些"问题"分子在PDB文件中都被标识为杂原子(HETATM), 这样可以使用sed命令轻松地移除它们
结构质量
对结构文件进行进一步检查以了解其质量是一个好习惯. 例如, 在晶体结构解析的精修过程中, 谷氨酰胺和天冬酰胺的酰胺基团的取向可能不正确; 组氨酸残基的质子化状态和侧链取向也可能存在问题. 维基百科列出了与蛋白有机氨基酸最相关的化学性质, 可做参考. 为了进行正确的结构验证, 可以利用一些程序和服务(如WHATIF). 本教程假定所用结构对模拟目的而言足够好, 不存在问题, 可直接用于准备体系.
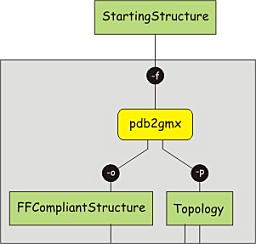
一个分子可由各个原子的坐标, 成键和非键相互作用的描述来定义. 由于从PDB文件获得的结构只含有原子坐标, 我们首先必须构建拓扑, 它给出了原子类型, 电荷, 成键情况等信息. 拓扑对应着某一特定的力场, 选择使用何种力场是一个值得仔细考虑的问题. 这里我们使用AMBER99sb-ILDN全原子力场, 它被广泛用于采样和折叠模拟的研究.
拓扑与结构的匹配很重要, 这意味着结构数据也需要根据所用力场进行转换. 可以使用pdb2gmx程序转换结构并建立拓扑数据. 该程序可以构建分子的拓扑, 前提是这些分子由特定的构建单元(如氨基酸)组成. pdb2gmx使用构建单元的数据库进行转换, 对于数据库中没有的分子或残基, 程序无法识别, 转换过程会失败. 输入下面的命令进行结构转换, 提示时选择AMBER99SB-ILDN力场和TIP3P(Transferable Intermolecular Potential 3P)水分子模型. 注意-ignh选项, 它表明程序会首先移除初始文件中的氢原子, 然后再根据相应力场的说明进行重建. 使用-ter选项后GROMACS会提示分子的末端变化, 合适的选择应该能够正确地表示体系. 在本教程中, 这取决于多肽是一个更大蛋白的片段(非带电末端)还是一个完整的分子(末端带电).
注意, 你可以使用下面的命令将gro文件和pdb文件进行互换
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> editconf<span style="color:#666666"> -f </span>XXXXX.gro<span style="color:#666666"> -o </span>XXXXX.pdb </pre></div>仔细阅读屏幕输出的提示, 并检查组氨酸质子化状态所做的选择, 注意蛋白质的总电荷数. 也要浏览输入结构的文件(protein.pdb)和输出的结构文件(protein.gro, GROMACS格式). 注意两种格式的区别. 最后浏览拓扑文件及其结构.
记下转换前后原子数目的区别, 解释为什么. 选择拓扑文件中的一个氨基酸残基, 列出其中的每个原子及其类型, 电荷.
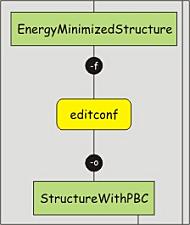
在对体系进行能量最小化前, 需要选定模拟体系的大致外形(空间排布方式). 大多数常见的模拟都是在周期性边界条件(PBC, periodic boundary conditions )下进行的, 这意味着要定义一个单位晶胞, 它可以填充堆积整个空间. 这样就可以模拟一个无限, 周期性的体系, 避免了由于模拟盒子的墙壁导致的边界效应. 只有少数几个通用形状可用于设置PBC. 我们将使用菱形正十二面体盒子, 因为它对应于球的最佳堆积, 因此对自由旋转的分子是最佳选择. 为避免周期映像之间的直接相互作用, 我们设置多肽和盒子边界之间的最小距离为1.2 nm, 这样两个相邻的堆积盒子的距离就会大于2.4 nm. 可以使用editconf命令来设置PBC:
看看editconf的输出, 注意体积的变化. 另外, 也看看protein-PBC.gro文件的最后一行(提示: 可使用命令tail -n 1 protein-PBC.gro). 在GROMACS格式(.gro)中, 最后一行指定单位晶胞的形状, 并总是使用三斜矩阵的表示方法, 其中前面的三个数字指定对角元素(xx, yy, zz), 后面的六个数字指定非对角元素(xy, xz, yx, yz, zx, zy).
新盒子的体积多大?
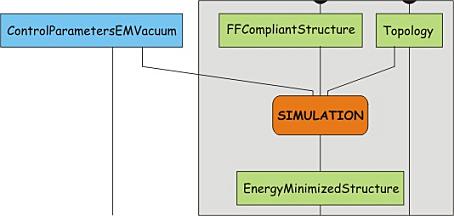
现在, 一个适合所选力场并且格式正确的结构文件已经建好了, 同时还得到了相应的拓扑和盒子. 然而, 由于结构转换中, 牵涉到氢原子的删除和添加, 这可能会导致局部有些过大的应力, 比如因两个原子的位置太接近而引起的作用力. 因此我们必须对结构进行能量最小化, 使其松弛一点. 这其实就是一个模拟步骤, 包括两个过程: 第一步, 结构和拓扑同时还有一些控制参数被整合在一起, 形成描述整个体系的单个文件. 这个过程会生成一个运行输入文件, 该文件可以作为唯一的输入文件, 用于第二步动力学模拟的mdrun程序. 把这个过程分为两步的好处是, 运行输入文件可以传送到专门的(高性能)计算机网络或者超级计算机, 并在那里完成模拟计算. 然而, 一般只在成品模拟中才会这么做.
为了执行这些步骤, 需要用到一个.mdp参数文件, 将该文件01_em_vac_PME.mdp保存到自己的工作目录下. 该文件包含一些能量最小化的控制参数, 请查看其中的内容. 注意, 文件中以integrator开始的行指定了所用的算法. 本例中, 指定采用最速下降法进行1000步能量最小化. 现在, 用以下命令整合结构和拓扑文件以及一些控制参数:
grompp程序将会提示此体系的净电荷不为零, 还会输出一些与体系和控制参数有关的其他信息. 该程序也会产生一个额外的输出文件(mdout.mdp), 里面包含所有控制参数的设置. 下一步, 运行mdrun
由于只有3150个原子, 能量最小化很快就完成了. 使用选项-v会将每步的势能和最大受力打印出来, 以便于跟踪能量最小化过程. -deffnm选项设定了所有输出文件的默认文件名, 避免了对每个输出文件进行命名, 从而减少了需要设置的选项. 现在, 结构弛豫好了, 我们该添加溶剂了.
使用了什么方法进行能量最小化? 参数文件中指定了多少步, 实际使用了多少步进行能量最小化? 什么原因致使能量最小化在指定步之前停止? 体系最终的势能为多少? 使用PyMOL载入能量最小化之前和之后的结构, 并进行比较.
为了更好地查看结构的区别, 可以将优化后的结构叠合到初始结构上(已经重命名为protein.pdb). 使用align命令可以将结构protein-EM-vacuum.pdb与protein.pdb对齐,
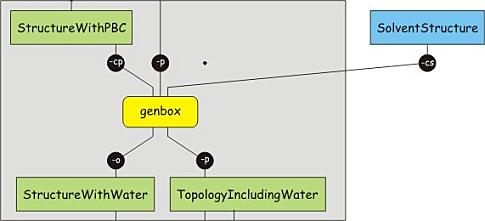
现在单位晶胞已经设定好了, 结构也优化好了, 我们可以添加溶剂了. 有好几种溶剂模型, 每种模型多少都与某一力场密切相关. Amber99SB力场通常使用TIP3P水分子模型, 这个模型你已经在前面选择过了. 添加溶剂不需要拓扑, 但可能需要更新拓扑, 在其中添加溶剂. 使用以下命令向盒子中添加TIP3P溶剂:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> solvate<span style="color:#666666"> -cp </span>protein-EM-vacuum.gro<span style="color:#666666"> -cs </span>spc216.gro<span style="color:#666666"> -p </span>protein.top<span style="color:#666666"> -o </span>protein-water.gro </pre></div>看看protein.top文件的结束部分, 其中有添加溶剂的内容, 检查添加的溶剂分子的数目.
向体系中添加了多少水分子, 对应的体积多大? 体系中现在有多少原子?
用以前学过的命令, 将.gro文件转换为.pdb文件. 用PyMOL程序载入溶剂化的结构(protein-water.pdb). 在PyMOL中, 可用下面的命令显示盒子形状
执行命令后, PyMOL会绘制一个线框来显示三斜晶胞盒子的边界. 可能看起来不太明显, 但这个三斜形状对应于菱形十二面体. 另一个值得注意的事情是, 并非所有的溶剂都处于盒子内, 但都以长方体块状排布. 有时, 蛋白质也会部分地伸出水外面.
蛋白伸出水面外部为什么不是问题?
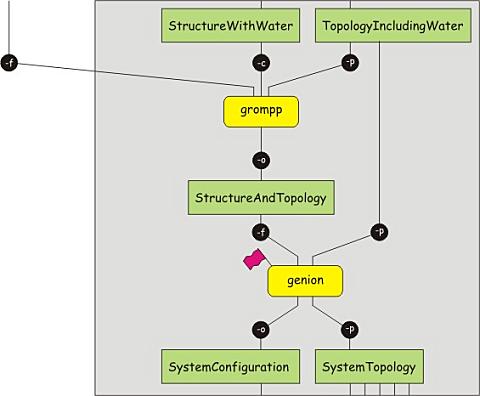
目前我们已经有了溶剂化的蛋白质, 但体系仍存在净电荷. 为了使体系呈电中性, 我们必须添加一定数目的抗衡离子. 此外, 添加一定浓度的离子, 可认为是一个好的做法. 程序genion能完成这些任务, 但它需要一个运行输入文件作为输入文件, 即一个包含了结构和拓扑的文件. 就像前面一样, 我们可以使用grompp来生成这样的文件. 下载新的mdp文件, 并执行下面的命令
接下来, 文件protein-water.tpr可作为genion程序的输入文件. 我们设定了NA+/CL-(-pname NA+ -nname CL-)的浓度为0.15 M(-conc 0.15), 并指定必须加入超量的某一特定种类的离子以使体系呈电中性(-neutral). genion程序会询问用离子取代何种分子, 选择SOL组.
注意: 为中和体系电荷而添加的离子的数量. 通过将一些水分子替换为离子, 体系的拓扑文件protein.top不再正确. 你可以修改拓扑文件, 减少溶剂分子的数目, 同时在文件的最后一部分(molecules)中SOL的后面增加一行, 指定添加的NA离子的数目, 并再增加另一行指明CL离子的数目. genion也提供了一个-p选项能够自动更新拓扑文件, 更方便.
向体系中添加了多少钠离子和氯离子?
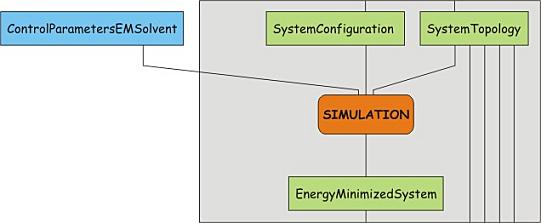
到此为止, 整个模拟系统已经准备好了. 在开始成品模拟前, 唯一要做的是对体系再次进行受控的弛豫. 由于添加溶剂以及替换离子, 可能会产生不利的相互作用, 例如, 原子间的重叠, 同种电荷太接近等, 因此需要对体系再次进行能量最小化, 其步骤与前面相同: 先运行grompp, 再运行mdrun:
参数文件中指定了多少步, 优化过程进行了多少步? 体系最终的势能多大?
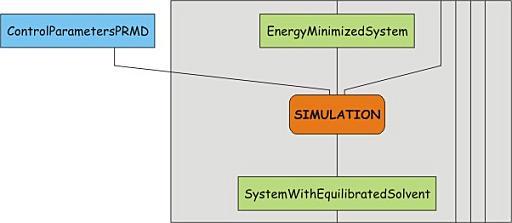
为了耗散掉体系的最大应力, 我们使溶剂分子适应蛋白, 即, 我们允许溶剂自由移动, 而将蛋白的非氢原子或多或少地固定在参考位置. 这么做的目的是为了确保溶剂的构象"匹配"蛋白质. 这一步骤是第一个真正的MD步骤, 控制参数在03_nvt_pr1000_PME.mdp文件中. 浏览这个文件, 注意integrator行和define行. 后者用于控制拓扑文件的内容: define = -DPOSRES将定义全局关键字POSRES. 查看拓扑文件最后的内容, 理解位置限制是如何打开的.
也要注意, 我们开始引入温度并对体系进行热浴耦合. 换句话说, 体系将在打开温度耦合的情况下运行一小段时间, 让体系弛豫到一个新的状态. 为此, 我们为粒子赋予了速度, 这是通过参数gen_vel来控制的, 并在下一行设定了速度分布(Maxwell分布)的温度和随机数生成器的种子(gen_seed). gen_seed被设为-1, 但应该更改为另外的值(提示: 可以使用命令更改, 如sed -i /^gen_seed/s/-1/设定值/ npt.mdp). MD模拟是最重要的一步, 所以每次都以相同的坐标, 速度和相同的参数开始模拟时, 就会导致模拟结果一模一样, 这不是我们所希望的.
查看上面提供的mdp文件, 你使用哪个温度模拟体系? 选择这一特定温度的原因何在?
编辑完参数文件后, 使用grompp和mdrun程序来准备和运行模拟:
为什么我们建议你修改随机数种子的值而不是使用自动生成的随机种子(默认值
-1)? 模拟长度是多少ps? 在拓扑文件中位置限制是如何包含进去的? 有两个分别耦合到热浴的组, 它们是哪些组, 每个组中包含哪些粒子?
看看总能量, 势能和动能的变化会很有趣. 在前一步, 粒子没有速度, 所以没有动能, 因此也没有温度. 现在, 模拟一开始, 原子就被赋予速度因此获得了动能.
模拟过程中的能量信息被保存在不可读的(二进制)文件中, 其扩展名为.edr. 该文件中的信息可用gmx energy命令提取. 绘制体系的温度, 势能, 动能和总能量随时间变化的图.
运行上面的命令后, 会提示你一系列能量项以供选择, 所选能量项的结果将会被写入指定的输出文件中. 输入与温度, 势能, 动能和总能量相对应的数字, 最后输入0(零), 回车. (提示: 你可以通过使用管道(|)自动选择某些项, 例如echo 10 11 12 14 0 | gmx energy ..., 或echo Temperature 0 | gmx ernergy ...)
输出文件(.xvg)可以用xmgr或xmgrace程序查看, 这些程序需要预先安装好. 也可以用我们提供的Python脚本xvg2ascii.py查看, 它能在终端窗口上显示出基于字符的图形.
注意: 绿色曲线是总能量, 黑色是势能, 蓝色是温度, 红色是动能. 你也可以点击曲线并填写Legend窗口以改变图例. 别忘了点击accept以接受改变.
温度如何变化? 势能/动能/总能量如何变化, 如何解释?
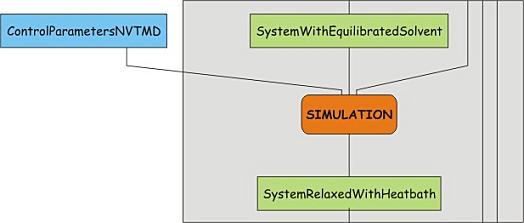
预平衡常常分为两个阶段, 第一个阶段使用NVT系综(粒子数, 体积, 温度恒定), 这一系综通常也称为等温等容系综或正则系综. 这一预平衡过程所需的时间步数取决于体系的性质, 但在NVT中, 体系的温度应该会在需要的值附近达到一个平台. 如果温度没有稳定. 就需要延长模拟时间. 作为示例, 我们在前面已经进行了这一步骤, 现在就进行下一步骤.
前面的NVT预平衡, 稳定了体系的温度. 在进行数据收集前, 我们也必须稳定体系的压力(因此也包括稳定密度). 压力平衡在NPT系综下进行, 这一系综的粒子数, 压力和温度保持不变, 也被称为等温等压系综, 最能代表实验条件. 现在开始慢慢放开限制, 让体系弛豫到新的状态. 下载参数控制文件04_npt_pr_PME.mdp, 看看其中的压力耦合参数. 然后使用grompp和mdrun运行模拟:
查看控制参数文件, 找到控制压力耦合的参数.
运行结束后, 再次查看一下能量和温度. 使用前面的方法抽取它们. 数据提取方法同前:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> energy<span style="color:#666666"> -f </span>protein-NPT-PR1000.edr<span style="color:#666666"> -o </span>thermodynamics-NPT-PR1000.xvg </pre></div>观察能量图.
温度如何变化? 势能/动能/总能量如何变化, 如何解释?
现在开始重复相同的平衡模拟, 并逐步放开限制
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">sed</span><span style="color:#666666"> -e </span>'s/1000 1000 1000/ 100 100 100/g' posre.itp > tmp.itp <span style="color:#AA22FF">mv</span> tmp.itp posre.itp <span style="color:#AA22FF">gmx</span> grompp<span style="color:#666666"> -v </span>-f 04_npt_pr_PME.mdp<span style="color:#666666"> -c </span>protein-NPT-PR1000.gro<span style="color:#666666"> -p </span>protein.top<span style="color:#666666"> -o </span>protein-NPT-PR100.tpr <span style="color:#AA22FF">gmx</span> mdrun<span style="color:#666666"> -v </span>-deffnm protein-NPT-PR100 </pre></div>再减小一次
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">sed</span><span style="color:#666666"> -e </span>'s/100 100 100/ 10 10 10/g' posre.itp > tmp.itp <span style="color:#AA22FF">mv</span> tmp.itp posre.itp <span style="color:#AA22FF">gmx</span> grompp<span style="color:#666666"> -v </span>-f 04_npt_pr_PME.mdp<span style="color:#666666"> -c </span>protein-NPT-PR100.gro<span style="color:#666666"> -p </span>protein.top<span style="color:#666666"> -o </span>protein-NPT-PR10.tpr <span style="color:#AA22FF">gmx</span> mdrun<span style="color:#666666"> -v </span>-deffnm protein-NPT-PR10 </pre></div>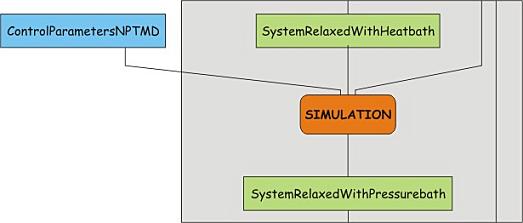
完成两个阶段的预平衡后, 体系在需要的压力与温度下平衡好了. 我们现在可以放开位置限制运行成品模拟以收集数据了. 过程与前面的类似, 因为我们要将检查点文件(在这种情况下它包含了压力耦合的信息)用于grompp. 下载文件05_npt_NOpr_PME.mdp.
运行结束后, 再次查看一下能量和温度, 同时注意观察压力变化. 使用前面的方法抽取它们. 数据提取方法同前:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> energy<span style="color:#666666"> -f </span>protein-NPT-noPR.edr<span style="color:#666666"> -o </span>thermodynamics-NPT-noPR.xvg </pre></div>看看能量, 温度和压力的变化曲线.
温度如何变化? 压力如何变化?
最后一个问题与如何从有限数量的粒子系统中提取热力学性质直接有关. 粗略地说, 热力学指的是大量粒子(例如, 几十亿个而不是几千个)的行为. 对大量粒子的性质进行平均能够减少波动, 相反, 仅对少量粒子进行平均, 较大的波动无可避免.
由于我们引入了压力耦合, 体系的密度会发生变化. 从能量文件中提取密度数据, 方法如下:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> eneconv<span style="color:#666666"> -f </span>protein-NPT-PR1000.edr protein-NPT-PR100.edr protein-NPT-PR10.edr protein-NPT-noPR.edr<span style="color:#666666"> -o </span>protein_equil_concat.edr <span style="color:#AA22FF">gmx</span> energy<span style="color:#666666"> -f </span>protein_equil_concat.edr<span style="color:#666666"> -o </span>density-NPT-noPR.xvg </pre></div>体系密度随时间如何变化? 如果打开压力耦合, 体系的密度为什么会变化?
终于到了最后一步. 我们已经得到了或多或少平衡好的溶剂化体系, 其中包含我们感兴趣的蛋白质, 所以该进行成品模拟了. 记住, 成品模拟并不表示整个模拟都可以用来分析感兴趣的性质. 虽然已经消除了初始构象的一些影响, 体系还不太可能已经达到平衡状态. 在分析阶段, 我们会检查模拟的哪一部分可以认为处于平衡状态, 适合用于进一步处理分析. 但首先需要设定运行参数. 这里只需要运行另一个模拟步骤, 类似于准备体系的最后一步. 然而, 这一步也是另一处需要考虑模拟目的的地方, 所以应当选择能满足待分析性质的有关控制参数. 可考虑以下问题:
我们将运行50纳秒的模拟. 下载控制参数文件06_md_PME.mdp, 先查看其中的内容.
为了完成50纳秒的模拟, 需要多少步?
你必须编辑参数文件, 指定运行步数使得总模拟时间为50 ns. 然后使用grompp命令将最终的结构文件和准备步骤中得到的拓扑文件合并为一个运行输入文件.
虽然运行输入文件是二进制格式, 我们还是可以查看其内容. 在某些情况下, 模拟会发生一些意外, 这可能与内部控制和力场参数有关. 在这种情况下, 查看运行输入文件尤为有用. gmx dump程序可以将运行输入文件转换为可读格式. 转换后的内容可能会有很多页, 因此建议将其重定向到一个文件中, 或使用more或less命令分屏显示文件. 输入以下命令查看运行输入文件的内容(注意, 在实际操作时, 需要把文件名protein_md.tpr替换为运行grompp后所得到的文件名):
模拟结束后就可以进行数据分析了. 这是一个重要的过程, 包括三个阶段. 首先, 有必要进行一些标准的检查, 以便对模拟质量进行评估. 如果评估结果表明模拟良好, 就可以对每个模拟进行分析, 回答预设的研究问题了. 最后, 来自不同模拟的结果可以综合起来.
注: 文件名应当能反映文件的内容, 根据模拟体系的不同而不同. 这里我们假定使用默认的文件名, 这意味着会有以下文件:
topol.tpr: 运行输入文件, 包含模拟开始时体系的完整描述confout.gro: 结构文件, 包含最后一步的坐标和速度traj.trr: 全精度轨迹, 包括随时间变化的位置, 速度和力traj.xtc: 压缩轨迹, 轻量, 只包含低精度(0.001 nm)的坐标信息ener.edr: 随时间变化的有关能量数据md.log: 模拟过程的日志, 包含模拟过程中的信息另外, 许多分析工具都能生成.xvg格式的文件. 这些文件能用xmgr或xmgrace程序查看, 也可用Python脚本xvg2ascii.py在终端以字符形式显示.
在进行其他分析之前, 必须确认模拟正常结束. 有许多原因可能导致模拟中断, 特别是涉及力场或体系平衡不充分的问题, 要检查模拟是否正常结束, 可以使用gmx check程序
确认模拟运行了50纳秒.
轨迹文件中有多少帧, 时间分辨率为多少?
模拟信息的另一个重要来源是是日志文件. 在md.log的结束部分给出了模拟过程的统计数据, 包括CPU和内存资源的使用情况以及模拟时间. 查看日志文件的结束部分. 如果你使用less, 可以按住G(shift-g)来略过前面部分, 跳到文件结束. (提示: 可以使用sed命令只抽取日志文件的最后部分: sed -ne '/NODE/,$p' md.log)
模拟实际运行了多少时间(小时), 模拟速度为多少(ns/day)? 要模拟1 s需要多少年? 势能的哪部分贡献消耗量大部分计算时间?
现在是有趣的部分, 尽管多数分析都归结为从轨迹文件中提取图像, MD当然最重要是的体系的运动. 我们要看看轨迹.
首先使用GROMACS提供的查看器ngmx来看看. 虽然该程序的完善程度和视觉效果不及其他查看软件, 但它能够根据拓扑文件的信息绘制成键. 其他查看软件可能隐含长程键, 导致这些键被认为太长而不画出, 或者会在非常接近的原子之间画出键. 这是对模拟结果分析的一个常见错误. 使用ngmx载入拓扑和轨迹文件:
看看程序菜单, 试试不同的选项. 播放动画. 观看过程. 通过右边的选项控制. 右击或左击选择选项来改变查看.
为了可视化轨迹, 我们将从轨迹中抽取1000帧(-dt 50), 并去除水分子(当提示选择时, 选择蛋白Protein). 此外, 我们还要消除跨过盒子边界的跳跃形成连续轨迹(-pbc nojump). 要处理这些事情, 我们使用瑞士军刀般的GROMACS工具trjconv, 它有1001个可能的组合选项. 我们使用它输出一个多模型的pdb文件, 用于PyMOL可视化.
当提示选择trjconv的输出组时, 选择蛋白Protein(组1)从而忽略溶剂与离子.(提示: 如前面所述, 可以使用echo来传递选项: echo 1 | trjconv ...)
在PyMOL中载入轨迹
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">pymol</span> protein.pdb </pre></div>载入所有帧后, 播放动画
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">mplay</span> </pre></div>当播放动画时, 所有其他控制仍可以工作. 你可以使用鼠标旋转或缩放体系, 也可以改变分子的表示模式.
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">spectrum</span> <span style="color:#AA22FF">show</span> cell </pre></div>如果多肽扩散超过盒子边界, 会发生什么?
如果一切正常的话, 现在你可以看到多肽的扩散, 翻转和扭动. 但我们对内部运动更感兴趣, 而不是整体行为. 在PyMOL中, 你可以使用命令intra_fit将轨迹中的所有其他帧与第一帧对齐, 随后, 你可以使用orient命令设定聚焦点在多肽上
现在所有帧都已经叠合好了, 你可以看到多肽的一些部分比另一些部分运动得更剧烈, 这种运动的差异在后面会进行定量化分析.
当然, 使用卡通模式表示多肽显示效果会更好:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">show</span> cartoon </pre></div>上面的命令会将碳骨架表示为粗管状, 而无法显示正确的二级结构元素, 因为.pdb文件中没有二级结构信息. PyMOL能够计算蛋白的二级结构, 但只计算一帧, 然后将结果应用到所有的帧. 例如, 下述命令可以计算第一帧的二级结构:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">dss</span> </pre></div>通过指定状态编号, 可以改变要计算的帧
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">dss</span> state=1000 </pre></div>最后, 让我们同时查看所有帧, 检查多肽的柔性和刚性区域
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">set</span> all_states=1 </pre></div>请随便摆弄PyMOL. 试着放大柔性或刚性区域, 并检查侧链的构象. 请使用ray和png命令制作图像, 即使浪费点(CPU)时间也不要紧(提示: 将场景输出为POV-Ray格式, 得到的图像可能更酷). 但记住, 如果图像的场景太过复杂, 可能会导致PyMOL的内置光线追踪器崩溃, 这种情况下, 你可以直接使用png保存屏幕上的图像.
如果有足够的机时, 你可以考虑制作一段不错的动画. 你可能已经注意到了, 轨迹的噪声很大. 这基本上是热噪声, 因此只是蛋白正常行为的一部分, 但这些噪声对制作好的动画会有影响. 我们可以滤除这些高频的运动, 只保留更慢更平滑的整体运动. 为此, 可以使用filter命令:
现在在PyMOL中载入滤过后的轨迹, 设置二级结构(dss), 显示二级结构(show cartoon), 隐藏碳骨架上的侧链(hide lines, not (name c,n,o)), 以你喜欢的颜色进行着色, 然后使用orient命令设置好视角. 现在可以开始制作动画了:
退出PyMOL(quit), 检查目录下的文件(ls), 你会发现多了好些文件, 包括250张图片. 以每秒30帧的速度, 这些图片可以制作大约8秒的动画. 下载mpeg_encode程序和参数文件movie.param, 用它来生成单帧图像的动画(你可能需要编辑参数文件来改变文件名):
对轨迹进行了最初的可视化检查后, 该对模拟质量进行一些更彻底的检查了. 质量保证(QA)包括对一些热力学参数收敛程度的测试, 如温度, 压力, 势能和动能等. 更常用的含义, QA试图评价模拟是否达到平衡. 通过起始结构和平均结构的均方根偏差(RSMD)也可以对结构的收敛性进行检查. 接下来必须检查相邻周期性映像之间没有相互作用, 因为这种相互作用会导致一些非物理效应. 最后一步QA测试是计算原子的均方根波动, 并与晶体学数据的b因子进行比较.
我们首先从能量文件中提取一些热力学数据, 包括温度, 压力, 势能, 动能, 晶胞体积, 密度以及盒子大小等. 这些量中的大多数已经在前面的准备步骤阶段检查过了. 能量分析使用energy命令进行, 该程序读入能量文件, 也就是模拟过程中生成的扩展名为.edr的文件. energy命令会提示需要从能量文件中抽取哪些项, 并为其生成一个图形. 使用下面的命令
执行命令后将列出一系列存储在.edr文件中的能量以及相关项. 本教程的能量文件中可能含有59项, 每一项都可以抽取并作图. 最前面的9项对应于力场中的不同能量项, 44以上的项列出对蛋白Protein和非蛋白Non-Protein组划分后的结果, 包括二者之间的相互作用. 键入要提取的性质的序号并回车就可以得到相应的.xvg数据文件. 要抽取温度, 键入temperature 0或12 0并回车(也可不键入0, 但需要回车两次). 使用xmgrace查看下图形, 观察温度如何围绕设定值(310 K)上下波动.
也可以从热力学性质的涨落计算体系的热容. 为此, 除体系温度外, 还必须从.edr能量文件中抽取焓(NPT系综)或总能量Etot(NVT系综)的值. 进一步, 我们必须使用-nmol选项明确指定体系中的分子数目(你可以查看拓扑文件的最后部分获知体系中分子的总数). 这样energy就可以自动计算热容并在输出部分的最后报告这个值. 更多细节请参考手册D.29.
体系的平均温度和热容多少?
通过名称引用能量项可实现能量文件的自动处理. 使用echo和管道(|)可以将一个程序输出重定向为另一个程序的输入, 这样energy的选择可以自动完成. 要抽取多个项, 各项之间必须以\n分割,
复制粘贴或键入以下命令行来抽取其他项.
逐个查看这些文件, 观察对应数值的收敛情况. 如果有的数值没有收敛, 这意味着模拟还没有达到热力学平衡状态, 必须延长模拟时间才能进行进一步的分析. 此外, 达到平衡前的过程是不能用于分析的. 这里, 为简单起见, 我们忽略这些考虑, 直接使用模拟的结果进行分析.
energy.xvg和box.xvg文件中给出的是什么性质? 估计压力, 体积和密度的稳定值.
一些性质收敛慢, 达到平衡所需的时间长于另一些性质. 特别的, 温度很容易收敛而体系各部分间的相互作用可能收敛较慢. 这样会导致温度已经收敛到平衡值, 而体系不同部分之间的相互作用仍需要更长时间进行平衡. 查看多肽与溶剂之间的相互作用能:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">echo</span> "48\n50 0" | gmx energy<span style="color:#666666"> -f </span>ener.edr<span style="color:#666666"> -o </span>coulomb-inter.xvg <span style="color:#AA22FF">echo</span> "49\n51 0" | gmx energy<span style="color:#666666"> -f </span>ener.edr<span style="color:#666666"> -o </span>vanderwaals-inter.xvg </pre></div>
coulomb-inter.xvg和vanderwaals-inter.xvg文件中给出的是什么量?
质量保证中最重要的检查事项之一就是确保周期性映像之间没有直接的相互作用. 由于周期性映像是全同的, 其间的相互作用是物理上不应该发生的自相互作用, 会导致模拟结果不合理. 设想具有偶极矩的蛋白会有直接的相互作用. 那么同一蛋白处于盒子边界的两个末端之间就会产生吸引, 这将影响蛋白质的自身的行为并导致模拟结果不合理. 我们通过计算每一时刻周期性映像之间的最小距离来证实这种情况不会发生. 这通过mindist来实现:
周期性映像之间的最小距离多大, 何时出现? 在你的模拟过程中, 用于长程非键作用的截断距离是多少? 根据这一距离, 两个周期性映像之间允许的最近距离多大? 当最小距离小于截断距离时, 哪一非键能量项受影响最大, 为什么? 如果最小距离小于截断距离, 会发生什么情况? 你的模拟中是否出现了这种情况? 选择
C-alpha组重新运行mindist, 结果有变化么? 对你的体系而言, 这意味着什么?
要注意小距离事件是不时出现还是持续出现. 如果持续出现, 很可能影响蛋白的动力学; 如果只是偶尔出现, 那基本没有影响.
不仅直接相互作用需要担心, 非直接效应, 即以水为媒介的间接相互作用也可能引起问题. 例如, 蛋白可以导致水在离其最近的四个溶剂化层中出现有序性, 这大约对应于1 nm的距离. 理想情况下, 最小距离不应该小于2 nm.
回旋半径取决于某(些)原子质量与分子重心的关系, 可用于表征蛋白质结构的密实度. 作为QA的一部分, 我们将计算回旋半径, 它给出了每一时刻分子形状的信息, 可以与实测得的水动力学半径相比. 可以使用gyrate计算回旋半径, 这一程序将会给出回旋半径的各个分量, 对应于惯性矩阵的本征值. 这意味着第一个单独的分量对应于分子的最长轴, 最后一个对应于最短轴. 实际上, 三个轴的值给出了分子形状的整体描述,
查看回旋半径及其分量, 注意这些值如何达到平衡值.
回旋半径收敛了么? 如果是, 什么时候收敛, 收敛值为多少? 不同多肽模拟给出的回旋半径其涨落行为是否类似, 为什么? 你能将回旋半径的涨落与周期性映像的最小距离联系起来么?
除了能量等性质, 也能够通过结构的变化和弛豫来考察模拟趋向平衡的收敛性. 通常, 这种弛豫仅仅使用结构到参考结构(如晶体结构)的欧几里德距离来衡量. 这一距离被称为均方根偏差(RMSD). 然而, 我们也建议再考察一下到平均结构的弛豫, 即相对于平均结构的RMSD, 个中原因将在下节说明. 但是要计算相对于平均结构的RMSD, 需要首先获得平均结构. 平均结构可以在计算均方根波动(RMSF)时顺便获得.
RMSF计算每个原子相对于其平均位置的涨落, 表征了结构的变化对时间的平均, 给出了蛋白各个区域柔性的表征, 对应于晶体学中的b因子(温度因子). 通常, 我们预期RMSF和温度因子类似, 这可以用于考察模拟结果是否与晶体结构符合. RMSF(和平均结构)使用rmsf命令计算. -oq选项可以计算b因子, 并将其添加到参考结构中. 我们最关心的是每个残基的涨落, 这可使用选项-res设定.
使用xmgrace查看RMSF的图形, 区分柔性和刚性区域.
确定最柔性区域的起始和终止残基编号, 给出其最大振幅. 比较不同多肽构象的结果. 是否有区别? 如果是, 哪个构象柔性最大, 哪个构象柔性最小?
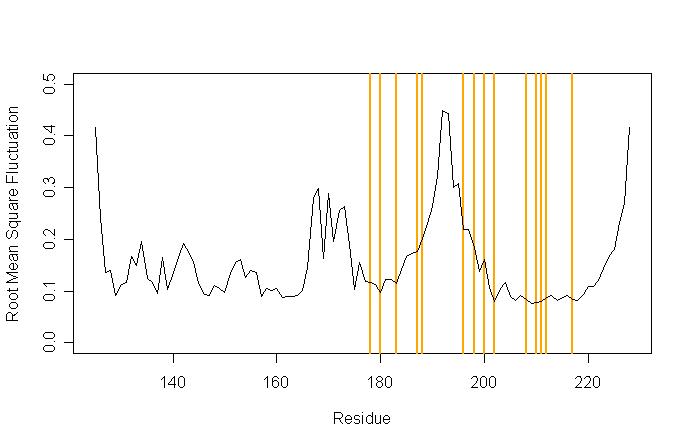
为了对这些结果获得关联性的印象, 这里有个人类朊蛋白质1qlz的RMSF, 图中标示出了会导致CJD疾病的突变残基.
将两个pdb文件载入PyMOL, 根据b因子对结构bfactors.pdb进行着色, 并确定柔性区域. 平均结构是非物理结构. 查看下其中的一些侧链, 注意观察平均对构象的影响.
下图是根据计算的b因子对1KLU多肽的参考结构进行着色后的结果(提示: 下图的设置如下: as sticks; show spheres; set stick_radius, 0.12; set sphere_scale, 0.25)

颜色分布在b因子值的范围内, 其中蓝色表示最小值(最稳定), 红色表示最高值(波动最大). 你可以通过截断最大值来调整颜色范围, 例如将其设置为350:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">Q</span> = 350; cmd.alter("all", "q = b > Q and Q or b"); spectrum q, selection=bfactors-residue </pre></div>如果你很好奇, 可以再次计算b因子, 但这次使用每个原子的值:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">g_rmsf</span><span style="color:#666666"> -f </span>traj.xtc<span style="color:#666666"> -s </span>topol.tpr<span style="color:#666666"> -o </span>rmsf-per-atom.xvg<span style="color:#666666"> -oq </span>bfactors-atom.pdb </pre></div>在当前的PyMOL中载入新生成的bfactors-atom.pdb文件, 这样可以直接将其与残基平均的b因子相比较. 如果需要, 不要忘了重新调整颜色范围.
比较和对照两个b因子的结构.
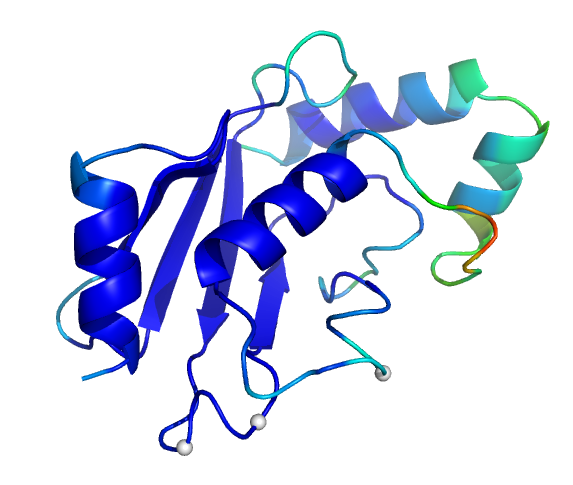
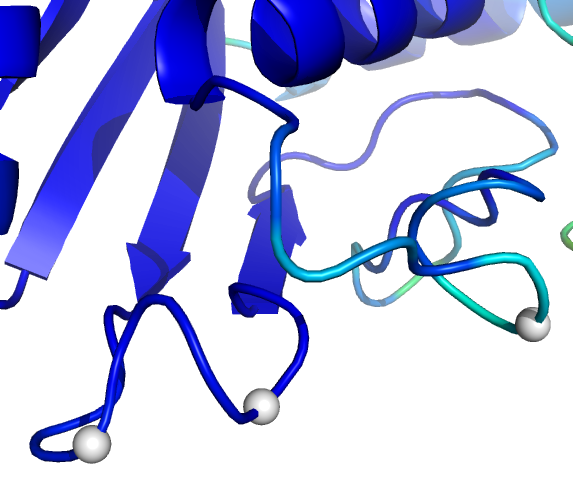
以下图像显示的是根据模拟计算得到人类野生型UbcH8蛋白的b因子着色图. 蓝色对应低值, 红色对应高值.白色区域表示目前已知的能够反转蛋白质相互作用特征的残基. 在图像右侧, 你可以看到那些在螺旋2的前后环区有较高的b因子.第二个图像是螺旋2的前环的放大显示.
当心! 由于你的多肽可能会跳出盒子外, 我们必须处理轨迹, 将粒子重新置于中心的周期性映像中. 为此, 可使用下面的命令:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> trjconv<span style="color:#666666"> -f </span>traj.xtc<span style="color:#666666"> -o </span>traj_nojump.xtc<span style="color:#666666"> -pbc </span>nojump<span style="color:#666666"> -dt </span>50 </pre></div>由于计算RMSF时也得到了平均结构, 我们现在可以计算均方根偏差(RMSD)). RMSD通常用于表征结构到平衡态的收敛情况. 如前面所讲, RMSD是结构变化对原子总数的平均, 基本上是一个距离表征, 低的值最有意义. RMSD可以用rms命令计算, 此命令可以实现对不同时刻不同分组的原子进行结构平均. 首先计算所有多肽原子的RMSD, 使用初始结构作为参考结构
如果观察到了, 在什么时间RMSD达到平台期, 平衡值多少?
再次计算, 但只考虑骨架原子:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> rms<span style="color:#666666"> -f </span>traj_nojump.xtc<span style="color:#666666"> -s </span>topol.tpr<span style="color:#666666"> -o </span>rmsd-backbone-vs-start.xvg </pre></div>这次的RMSD值更低, 这是由于计算时排除了通常更柔性的侧链原子, 在两种情形下. 两个RMSD都应该增大到一个平台值, 这意味着相对于参考结构, 多肽的结构达到了一定的距离, 然后或多或少保持在那个距离. 然而, 随着距离的增加, 可能的构象数目也在增加, 这意味着尽管RMSD达到了一个平台值, 但结构可能仍在趋近于其平衡态. 为此, 我们建议同时也检查趋向于平均结构的收敛情况.
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">echo</span> 1 | gmx trjconv<span style="color:#666666"> -f </span>traj_nojump.xtc<span style="color:#666666"> -s </span>topol.tpr<span style="color:#666666"> -o </span>traj_protein_nojump.xtc <span style="color:#AA22FF">gmx</span> rms<span style="color:#666666"> -f </span>traj_protein_nojump.xtc<span style="color:#666666"> -s </span>average.pdb<span style="color:#666666"> -o </span>rmsd-all-atom-vs-average.xvg <span style="color:#AA22FF">gmx</span> rms<span style="color:#666666"> -f </span>traj_protein_nojump.xtc<span style="color:#666666"> -s </span>average.pdb<span style="color:#666666"> -o </span>rmsd-backbone-vs-average.xvg </pre></div>与前面得到的图像进行比较. 注意在哪一点RMSD值开始趋平.
简要讨论下两个(相对于起始结构与相对于平均结构)图像的区别, 哪一个更能表征收敛情况?
到从为止, 我们完成了分析的第一部分, 包括可视化考察和质量确保检查. 现在该深入一点, 发掘一下蛋白质内部的情况. 分析的第二部分包括可根据多肽构象计算的结构性质, 例如氢键数量, 溶剂可及表面或特定的原子-原子间距离等.
说明:
Protein组.xmgrace可以使用滑动平均方法对数据进行平滑(去除噪声): Data -> Transformation.确认模拟已经收敛到平衡态后, 就可以进行一些真正的分析了, 模拟数据的分析可以分为几种类型. 第一类包括对单个构象进行解释, 在每个时间点根据一些函数获得一个值或多个值. RMSD和回旋半径就是例子. 这样的性质, 可称为构象依赖或瞬时性质. 此外, 也可以在时域对过程进行分析, 例如, 通过对一段时间内的平均化得到(自)相关或涨落. 在本部分会进行一些常见的分析, 其中的每种分析都会得到直接由轨迹(随时间变化的坐标)导出的某个值的时间序列. 问题可以参考程序运行时的屏幕输出或图像.
氢键数目是一类信息丰富的性质, 无论是内部氢键(蛋白-蛋白)还是蛋白和周围溶剂之间的氢键. 氢键存在与否可以通过氢键施体-H-受体之间的距离和施体-H-受体之间角度来推断. hbond命令可计算模拟过程中分子间或组间的氢键数目以及氢键距离或角度的分布. 使用下面的命令, 然后查看得到的输出文件
讨论两种情形下氢键数目的关系, 每种情形下的氢键数目的波动情况.
特定的氢键可以使用包含待研究原子编号的索引文件来考察. 查看多肽前一半和后一半所涉及的氢键. 你必须看看confout.gro文件来检查残基编号并将多肽大致分为两半. 假定你的多肽含有14个残基, 其编号始于22终于35. 你想将它分为两部分, 22-28和29-35. 下面的第一个命令将在组选择菜单中创建两个新的条目, part_1和part_2. 第二个命令是一个使用索引文件运行hbond的通用命令. 你可能想要看看N端和C端之间的氢键, 例如, 可以用来监测β发卡的形成.
注意, 你必须根据自己的多肽替换上面命令中的数字.
根据氢键分析, 你的多肽在模拟过程中是否形成类似β发卡的构象?
判定蛋白结构的最常用参数是指定二级结构元素, 如α螺旋, β片层. 能提供这一信息的一个程序是dssp, 但前提是你的电脑已经安装了dssp程序. 此程序并不是GROMACS发行的一部分, 但能够从CMBI, Radboud University获得. 下载后请解压, 然后将环境变量DSSP设定为程序的路径, 如/home/student/dssp.
GROMACS提供了一个dssp的接口, 可以计算轨迹中每帧的二级结构.
首先, 需要生成消除跳跃的轨迹
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> trjconv<span style="color:#666666"> -f </span>traj.xtc<span style="color:#666666"> -o </span>traj_nojump.xtc<span style="color:#666666"> -pbc </span>nojump<span style="color:#666666"> -dt </span>50 </pre></div>然后运行以下命令
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> do_dssp<span style="color:#666666"> -f </span>traj_nojump.xtc<span style="color:#666666"> -s </span>topol.tpr<span style="color:#666666"> -o </span>secondary-structure.xpm<span style="color:#666666"> -sc </span>secondary-structure.xvg </pre></div>secondary-structure.xvg文件包含一个时间序列, 列出了每帧中与每一二级结构类型相关的残基数目. 更多的细节在.xpm文件中, 它使用颜色编码了每个残基在一段时间内的二级结构. .xpm文件可以使用类似Gimp的程序查看, 但可以使用GROMACS工具xpm2ps添加一些有用的元数据, 得到的结果可以使用gview查看, 或转换为pdf后使用xpdf或evine查看.
讨论二级结构的变化, 如果有的话. 比较不同蛋白二级结构的稳定性.
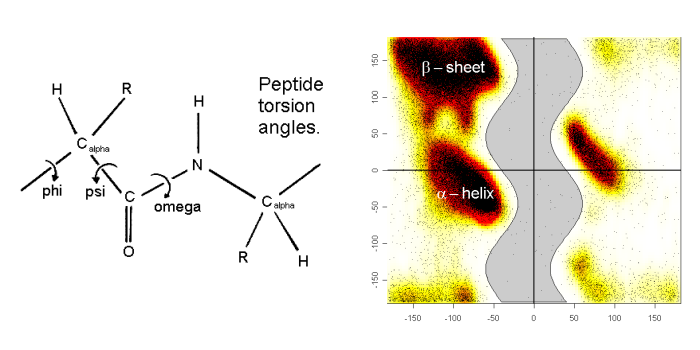
蛋白质骨架的phi和psi扭转角是两个洞察蛋白质结构特性的有用参数. phi对psi的绘图称为拉氏图, 该图中某些特定区域反映了蛋白二级结构元素或氨基酸的特性, 而(这些区域之外的)其他区域被认为是禁阻的(不可到达). phi/psi随时间的变化可以体现出结构的转变. 这些角度可以通过rama程序进行计算, 尽管结果有些粗略, 因为程序将所有角度都输出到单一的文件中. 若想研究单个残基, 可通过Linux程序grep将其从图中选出来.
此文件中包含了全部氨基酸的所有phi/psi角. 要抽取某个残基的角度, 例如LEU-24, 可键入
抽取每个残基(除边界外)的拉氏图, 并用xmgrace对其进行可视化, 描述它们的主要相似性与差别.
根据拉氏图, 讨论每个残基的构象稳定性.
每组学生模拟了一个从不同构象开始的多肽, 这样做的目的在于增加采样, 或者增加模拟能够覆盖的结构空间. 为了对轨迹和多肽构象性质有一个完整的观点, 我们必须将轨迹拼合到一起.
由于大多数操作/计算在执行时都需要一个拓扑文件, 为避免使用哪个文件, 从那个轨迹开始之类的问题, 请从这里下载你的多肽文件. 这一文件包含了多肽分子位于原始蛋白-多肽复合物中的直角坐标. 你也可以使用这一结构来计算相对于结合结构的结构相似性. 下载pdb文件后, 你必须将其转换为GROMACS的坐标文件(.gro), 这是教程中第一步骤的重复操作. 别忘了选择合适的力场和水模型, 尽管后一选择对分析并无影响.
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> pdb2gmx<span style="color:#666666"> -f </span>XXXX_peptide.pdb<span style="color:#666666"> -o </span>XXXX_peptide.gro<span style="color:#666666"> -ignh </span></pre></div>这样, 使用新生成的这个文件来创建索引文件, 这样我们可以进行更多的选择. 在这一步, 与前面不同, 我们不需要指定任何特定的残基组, 因此, 我们简单地推出程序(键入q)
现在我们可以对我们新生成的轨迹进行截断, 使用trjconv去除前10 ns. -b选项设定要创建的新xtc文件的开始时间, 单位为ps, 这意味着我们需要新文件从10 ps开始. -dt选项指定我们要保留的时间精度. 当提示选择时, 选择Protein来生成xtc文件.
为什么我们只分析最后40 ns的轨迹?
下面的命令会输出一个单一的.xtc文件, 其中包含以下列顺序排列的四个轨迹, bound-helix-polypro-extended. 这一顺序很重要因为后面要进行比较, 因此需要特别注意. -settime选项指定在拼合轨迹时使用连续的时间, 这意味着首帧时间为10 ns的第二个轨迹, 会恰好放在第一个轨迹的后面50 ns处. 如果你忘记了这个选项, 所拼合的轨迹具有重复的时间戳(即每一轨迹的原点都是10 ns). 我们要选择选项congtinue或c, 指示trjcat从前一轨迹的最后时间戳处开始新的轨迹.
我们在前面已经计算过RMSD, 并用其来检查模拟的收敛性, 但它也可以用于更进一步的分析. RMSD是两个结构之间的表征. 如果我们对轨迹文件中每一对结构的组合计算RMSD, 就可以看到是否有属于同一类型或具有相同特征的结构组. 属于同一组的结构其RMSD值较低, 而与其他组结构的RMSD值更高. 利用矩阵来表示RMSD值, 可以用于识别转变状态.
要建立RMSD矩阵, 可使用rms处理两条轨迹. 如果你要单独考虑组(簇)及其在不同轨迹之间的转变, 可以将所有的四条轨迹合并为一条, 再使用rms生成交叉RMSD矩阵. 所有的RMSD计算时, 选择主链组Mainchain.
得到的矩阵是灰阶图. 为了显示更清楚, 可以使用彩虹梯度图.
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> xpm2ps<span style="color:#666666"> -f </span>rmsd-matrix.xpm<span style="color:#666666"> -o </span>rmsd-matrix.eps<span style="color:#666666"> -rainbow </span>blue <span style="color:#AA22FF">ps2pdf</span> rmsd-matrix.eps <span style="color:#AA22FF">evince</span> rmsd-matrix.pdf </pre></div>分析时为什么选择主链组? 你看到多少簇? 在不同的轨迹中, 你是否对相同构象进行了采样? 你是如何发现的? 什么是轨迹的重叠, 即对相同构象空间进行采样? 你能发现多少转变? 从这个分析你能得到什么结论? 这是你预期的结果么? 请证实你的观点.
基于结构间的距离RMSD, 可以将结构归并为反映构象可及性范围及其相对权重的一组组团簇. 这可以通过聚类算法来完成, cluster命令实现了一些聚类算法. 这一程序会生成好几个输出文件. 检查该程序的帮助文档, 了解每一个它们的含义, 然后运行程序. 注意, 我们已经计算了RMSD矩阵, 可以将它作为cluster的输入.
聚类算法使用的RMSD截断值为多大? 这一值代表什么, 如何影响得到的团簇数目? 共有多少团簇? 最大的两个其尺寸多大?
更改RMSD的截断值, 以获得适当数目的团簇(10到100之间).
使用PyMOL打开cluater.pdb, 比较前两个团簇的结构.
前两个团簇之间是否存在可觉察的差别?
采用RMSD比较结构的一个不足之处在于, 它包含了最小二乘叠合, 这会影响结果. 但是, 一个蛋白的结构也可以使用一系列的原子间距来表示. 这可以用于获得一个比较的表征量, 并且不依赖于叠合, 这就是距离RMSD(dRMSD). 可以利用rmsdist命令来计算dRMSD.
dRMSD何时收敛? 收敛值多大? 所得图像与标准RMSD相比有何区别? 查阅GROMACS手册, 看二者是如何计算的. 试着根据二者的计算方法解释你观察到的区别.
我们现在要回答构象采样的问题, 在我们的轨迹中, 我们是否对多肽的结合构象进行了采样? 为此, 我们要对多肽的结合构象再次运行rms. 我们可以使用合并的轨迹来进行这一分析, 而不是计算并生成四个不同的RMSD图形. 记住你生成的合并轨迹中构象的顺序, 这很重要. 当叠合计算RMSD值时, 选择主链Mainchain组.
如果在计算时我们包含整个蛋白(即所有原子), 而不是只选择主链
Mainchain原子, 结果会有什么变化? RMSD和距离RMSD图形有区别么? 在不同轨迹中, 你是否对多肽的结合构象进行了采样? 对你研究的情况, 关于对蛋白多肽识别的构型采样假说你能得出什么结论?
本分析的最后一步是计算自由能形貌(FEL, free energy landscape), 它由不同多肽构象开始的轨迹采样所得. 除了看起来很酷以外, 它能显示出 在整个模拟过程中多肽所经历的自由能的谷和丘. 此外, 在对接计算中需要选择有代表性的结构时, FEL会变得更有意义.
FEL表示一个映射, 分子在模拟过程中所经历的所有可能构象到相应能量的映射. 典型的能量是Gibbs自由能. 正如你所想象的, 使用直角坐标来代表不同的构象是不合适的(你能想象4维形貌么?). 因此, FEL通常使用两个变量来表示, 它们反映了体系的特定性质, 并表征了构象变化. 例如你可以使用绕一根特定键的扭转角, 或分子的回旋半径, 或相对于天然状态的RMSD来作为这两个变量. 第三个变量是自由能, 可以从体系相对前面所选变量的分布(概率分布)来估计. 当使用三维表示时, 形貌图中的谷表示低自由能区域, 代表体系的亚稳定构象, 丘表示连接这些亚稳定状态的能量势垒.
FEL的计算可以使用GROMACS命令sham, 为了体验更好, 我们提供了一些额外的脚本将FEL载入Mathmatica(建议使用版本9), 并漂亮地显示出来. 此外, Mathmatica还可以用于识别形貌中的谷, 并确定哪些坐标匹配这些特定的状态. 利用这些信息可以追溯sham的输入, 用于提取轨迹中的时间帧, 进而得到代表性的结构.
我们将计算两个变量用于表示自由能. 为表示多肽的构象变化, 我们计算其回旋半径, 以及相对于平均结构的RMSD. 我们不使用天然状态, 因为那会破坏后面的对接模拟. 并且, 我们可以基于合并的轨迹计算这些变量. 计算FEL的一个前提是充分采样, 或者使用不同的初始速度重复进行长时间的模拟, 或者如在我们所做的, 从不同的构象开始进行模拟.
我们如何从MD轨迹中抽取代表性结构用于对接模拟? 给一个前面用来分析多肽构象变化的方法, 并说明这种方法可用于此目的.
因为FEL分析依赖于在多肽构象空间的充分采样, 并基于构象的分布进行Gibbs自由能估计, 我们需要准备新的轨迹文件(.xtc), 其中的帧数是我们到目前为止所用的十倍. 注意我们没有使用-dt选项. 对所有四条原始轨迹重复下面的步骤, 注意要更改名字.
使用和前面完全一样的做法合并高分辨率轨迹(当提示时间戳时选择c或continue)
现在我们需要生成FEL计算所需的数据. 先计算合并轨迹的回旋半径Rg, 方法如前:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> gyrate<span style="color:#666666"> -f </span>traj_concat_10-50ns-highresolution.xtc<span style="color:#666666"> -s </span>XXXX_peptide.gro<span style="color:#666666"> -o </span>rg-concatenated_traj.xvg </pre></div>为计算相对于平均结构的RMSD, 首先必须重新计算整条轨迹的平均结构. 再次使用rmsf命令生成平均结构(-ox选项), 然后使用平均结构做参考, 利用rms计算RMSD, 方法如前. 当提示时, 对叠合和计算都选择蛋白Protein组.
sham命令需要一个文件, 其中包含多个列, 每一列代表不同的坐标. 为生成一个正确的输入文件, 我们使用Perl脚本sham.pl, 并将我们刚生成的两个xvg文件作为它的输入. Perl是一种很类似Python的编程语言. 这个脚本将两个文件作为输入, 并假定数据处于文件的列中. xvg文件的第一列通常是时间, 第二列是感兴趣的坐标. 与Python一样, Perl的计数也是从零开始, 因此选择列数时小心. 下载脚本sham.pl并执行下面的命令. 输出文件只是两个xvg文件的简单合并, 第一列代表时间, 第二列和第三列是特定时刻的Rg值和相对于平均结构的RMSD.
现在我们有了正确的sham输入文件, 可用于生成FEL. 如果你还记得, Gibbs自由能可以根据分布概率计算出来, 并且依赖于指定的温度. 使用sham的选项-tsham来指定正确的体系温度(如果你不记得这个值, 可检查成品模拟所用mdp文件中的值).
如果使用
sham计算FEL时, 指定非常高的温度, 会出现什么情况?
sham的输出可以使用xpm2ps输出到ps文件, 再用pdf阅读器查看. 但是, 当选择代表性结构时这种二维等值线图帮助不大, 需要查看数据时也很麻烦. 为此, 我们准备了一个Mathmatica文件, 可用于FEL的2D或3D可视化和检查. 由于Mathmatica不支持xpm文件, 我们准备了一个Python脚本用于将任意的xpm文件转换为3列数据的文本文件, 这样更易操控. 你可以下载脚本xpm2txt.py, 用它转换xpm文件
下载Mahmatica脚本, 然后打开, 遵照其中说明, 更改开头的文件路径. 如果一切正常, 你可以看到类似下面的图像
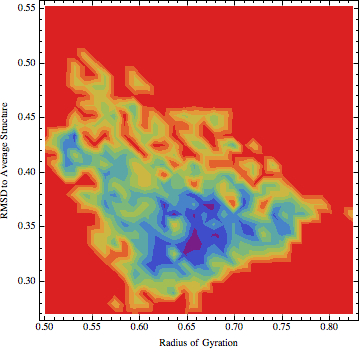
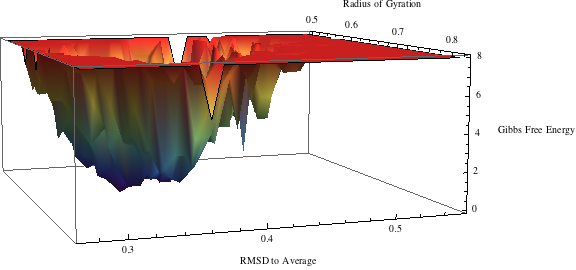
查看FEL并找到其最小点(谷)的位置. 选择你认为具有代表性的一些最小点(5). 你可以通过在2D等值线图上右键点击, 选择Get Coordinates选项来获知对应的坐标. 之后, 当你在图像上移动鼠标时, 会显示每个特定点的坐标. 记录这些坐标, 然后返回原始的sham.pl脚本输出文件查找相应的时间戳. 使用sham生成FEL的过程涉及坐标的分格, 因此对原始值有所近似, 不要期待你能找到精确的对应值.
从FEL中选择你认为能代表多肽状态的五个点, 记下它们的坐标.
为了在150000行中查找特定的内容(尽管使用gedit的搜索选项很方便), 我们提供了一个搜索时间戳的脚本. 可以使用它来得到5个时间戳. 你需要提供sham.pl的输出, 并给出两个坐标(顺序要正确). 下面是一个示例
如果你总是得到No timestamp found...这样的错误信息, 试着使用附近稍有区别的点, 或打开脚本增加变量nval的值到100或更大.
将代表性结构的时间帧与以前根据RMSD矩阵获得的相比, 这些代表性结构是否与结构簇相符?
一旦你有了5个点的时间戳, 你可以使用它们从合并的轨迹中抽取代表性结构. 下面的例子抽取45 ns时刻的结构:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">gmx</span> trjconv<span style="color:#666666"> -f </span>traj_concat_10-50ns-highresolution.xtc<span style="color:#666666"> -s </span>XXXX_peptide.gro<span style="color:#666666"> -dump </span>45000<span style="color:#666666"> -o </span>representative_45000ps.pdb </pre></div>在PyMOL中打开代表性结构以及结合结构, 并比较二者.
以下分析整理自泛素耦合酶教程, 用于说明操作过程, 但具体问题未必适用于前面的多肽-蛋白教程.
特定氢键可以用包含相应原子数量的索引文件来得出. 从分析1得出的RMSF及b-factors显示loops 2 和 3 (helix 2附近)值比较高. 实验数据也显示, loop 1可能在UbcH6 and UbcH8的多个行为中起了一定作用. 看看这些loop所包含的氢键连接. 第一个命令会在菜单中弹出3个新的条目去选择基团, 每个loop一个. 第二个命令是一个通用命令g_hbond, 需要一个索引文件. 你可能想看看每个loop里的氢键、loop1和loop2之间的氢键;例如, 某个特定的loop和蛋白质其他部分的氢键(为此可能需要修改索引文件)或者和水形成的氢键, 等...
除了氢键之外, 蛋白质不同的带电残基之间也常形成盐桥. 它对蛋白质的结构起着重要的稳定作用, 尤其是当它们处于憎水环境中时, 例如蛋白质核心. 但是盐桥也能在蛋白质暴露的表面形成, 这对于介导蛋白质的识别过程往往很重要. 残基间的盐桥分析可以用saltbr命令进行. 程序会输出一系列xvg文件, 给出-/-, +/-(最关注的)和+/+残基间的距离. 当需要时, 通过设置-sep选项, 这个程序可以为每对相反的带电残基产生一个输出文件, 这些残基位于轨迹中的某点, 彼此处于一定的截断距离范围内(这里是0.5 nm, 通过-t选项设置). 这将产生许多文件, 所以分析时最好建立一个单独的目录. 执行以下命令:
为了更清晰, 删除与钠离子和氯离子有关的文件:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">rm</span><span style="color:#666666"> -f </span>*CL-* *NA+* \#* </pre></div>看看以下残基之间的相互作用:
对于这些相互作用你有什么想法? 残基K60和D88高度保守, 仅仅将UbcH8中的残基D56突变为E56都会改变蛋白的相互作用方式. 其可能原因何在?
一个可能感兴趣的性质是溶剂可触及的蛋白质表面的面积, 通常称为溶剂可及表面(SAS, solvent accessible surface)或溶剂可及表面面积(SASA, solvent accessible surface area). 溶剂可及表面积可还可进一步细分为亲水性SAS和疏水性SAS. 此外, 结合一些经验参数, SAS还可以用于估计溶剂化自由能. 这四个参数都可用sasa命令计算, 它还可以计算每个残基或原子一段时间内的平均SAS. 输键入下面的命令, 要计算SAS的组和输出组都选择蛋白Protein, 然后查看输出文件.
哪个残基是最容易被溶剂触及的?
前面用到的rmsdist命令也可用于进一步的距离分析. 特别地, 为了解结构及其稳定性, 查看原子间的平均距离及其波动可能会有用处. 使用下面的命令对蛋白质中每对原子间的平均距离及其波动进行计算获得矩阵, 然后用上面的步骤重新着色, 并显示.png文件的结果(rmsmean.png和rmsdist.png).
简单地解释两个图像:
rmsmean表示结构,rmsdist表示柔性/稳定性. 回想前面分析得到的信息并查看结构.
从实验角度看这些距离同样重要. 原子间距离的边界值可以由NMR实验推断——利用核的Overhauser效应(NOE)——这也是NMR结构计算间的主要推动力. 如果蛋白质模型正确, 这个预期得可以到的NOE信号可通过MD模拟来计算. 这些信号与距离密切相关, 特别是r<sup>-3</sup>和r<sup>-6</sup>权重的距离. 这些信号也可以用rmsdist计算.
给nmr3.xpm和nmr6.xpm重新着色, 查看这些矩阵, 也查看noe.dat文件.
模拟战, 最小的1/r<sup>3</sup>和1/r<sup>6</sup>平均距离为多少?
计算向量的弛豫计算及其自相关. 对蛋白质, 通常包括碳骨架N-H或侧链C-H向量. 这种自相关给出了向量能保持其方向多长时间的量度, 因而为表征可变性和稳定性提供了指示. 序参量是自相关的长程限制. 如果一个分子能够自由旋转, 序参量将不可避免地减小到零; 但在分子框架内(内部参考框架), 序参量常有一个明确的值, 这个值表明总体稳定性. 通过叠合蛋白质, 这个参考框架可以固定下来, 因此序参量就确定了.
N-H序参量可以用chi命令计算. 这个程序可以写出一个.pdb文件, 把序参量加入了b因子列, 更容易查看. 该程序也计算J耦合参数, 并可以和NMR结果相比较, 或用于指导NMR实验.
看看.xvg文件中的序参量, 并用PyMOL查看.pdb文件, 根据b因子的值给残基着色.
记下起始与终止的残基, 具有最高序参量值的两个区域的平均值. 序参量与波动(RMSF)相比如何?
一个常用的, 但常常理解不深的分析方法是对轨迹进行主成分分析(PCA, PrincipalComponents Analysis). 这种方法有时候也称为本征动力学(ED, essential dynamics), 目的在于识别原子的大尺度集约运动, 从而揭示隐藏在原子波动后面的结构信息, 帮助确定哪些运动方式对蛋白质的整体动力学贡献最大.
在含有N个原子的体系中, 存在3N-6个可能的内部运动方式(另外6个自由度用来描述体系的外部整体的平动和转动). 在MD模拟中, 粒子的波动是互相关联的, 因为粒子彼此之间存在相互作用. 关联的程度有大有小, 但那些通过键直接相连或者彼此位置接近的粒子会产生显著的运动一致性. 粒子运动之间的相关性导致了体系总体波动的结构性, 对大分子而言, 这种结构常常与其功能或(生物)物理特性直接有关. 因此, 研究原子波动的结构性可以为了解这些大分子的行为提供有价值的洞见. 然而, 确实需要一定程度的线性代数方法和多元统计方面的知识才能来解释结果并认识到该方法的缺陷. 特别的, PCA的目标是用新的变量来描述原始数据, 这些新变量是原始变量的线性组合. 这也是PCA存在的最主要问题: 它仅仅使用原子运动间的线性关系来解释问题.
PCA的第一步是构建协方差矩阵, 它表征了每对原子之间原子运动的共线性程度. 协方差矩阵从定义上说是一个对称矩阵. 接着将这个矩阵对角化, 得到一个特征向量矩阵和特征值的对角矩阵. 每个特征向量描述了粒子的集约运动, 其中的向量值表示相应原子参与运动的程度. 通常, 体系中的大多数(>90%)运动是由10个以下的特征向量或主成分来描述的. 与特征向量相应的特征值等于以集约运动中的每个原子描述的波动的总和, 因此是与特征向量相关的总运动的一个度量, 可用于比较不同情况下蛋白质的可变性, 但对不同大小的蛋白质进行比较时, 就难以得到有意义的解释. 更多信息请参考Leach的9.14.
协方差分析会生成很多文件, 因此最好在一个新的目录中运行:
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">mkdir</span> COVAR <span style="color:#AA22FF">cd</span> COVAR </pre></div>协方差矩阵的构建和对角化可使用covar命令. 键入下述命令进行分析:
对PCA, 我们主要关心蛋白质骨架上的原子, 因此提示时选择backbone组. 构建和对角化协方差矩阵可能需要一些时间.
协方差矩阵的维数多少? 特征值的总和多少?
现在, 看看covar.xpm文件中的协方差矩阵.
矩阵显示了原子间的协方差. 红色表示两个原子运动一致, 而蓝色表示它们彼此向相反的方向运动. 红色的深度表示波动振幅的大小. 对角线上的值对应于前面得到的RMSF图.
查看除端基外运动最剧烈的两个部分, 它们之间如何相对运动, 它们各自相对于蛋白的其他部分如何运动?
从协方差矩阵能得到一组组相关或反相关运动的原子, 从而可以将其集约运动重新写入总运动. 我们前面提到过, 特征值保存在eigenvalues.xvg文件中, 通过相应特征值表示出总波动.
使用文本编辑器查看
eigenvalues.xvg文件, 计算头五个特征向量对总运动的百分比以及累积百分比.
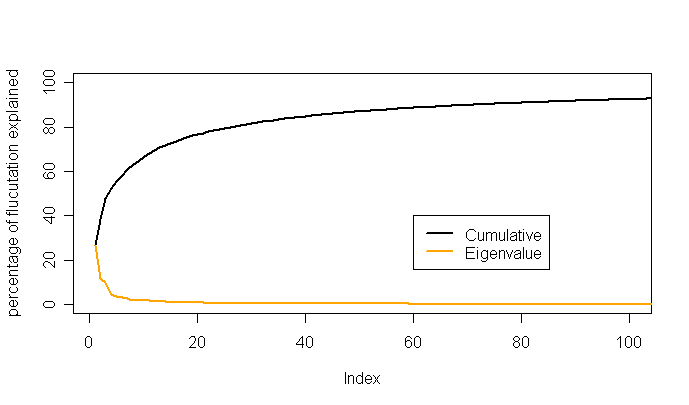
典型地, 最初五个特征值将捕获主要运动, 这相当于>80%的总运动. 如果解释的总波动较低, 就说明没有明确的集约运动.
为了解特征值的实际意义, 可用anaeig命令作进一步的分析. 为了更近地看看前两个特征值, 键入以下命令
特征值对应于运动方向. 选项-extr沿着选定的特征值从轨迹中提取极端结构. 把这些结构导入PyMOL查看:
把 pdf 文件中的模型分开, 删除原始结构.
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">split_states</span> ev1 <span style="color:#AA22FF">split_states</span> ev2 <span style="color:#AA22FF">delete</span> ev1 or ev2 </pre></div>给模型着色. 特征值1中的极端结构显示为蓝-绿色而特征值2为黄-红色.
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">spectrum</span> count <span style="color:#AA22FF">dss</span> <span style="color:#AA22FF">hide</span> everything <span style="color:#AA22FF">show</span> cartoon </pre></div>用PyMOL的align命令, 能画出表示两种构象差异的小条.
对特征值1而言, 极端结构之间的最大区别是什么? 对特征值2呢?
为了理解特征值的意义, 想象一下旅行推销员在欧洲城市间的移动. 这种移动可用地球坐标系统来说明, 对每个位置需要采用三个坐标. 虽然这样做没有问题, 但如果你只想解释推销员的移动, 这种方法不是最佳的. 因为理论上, 任何坐标系统都一样好, 我们可以定义一个新的坐标系统来解释推销员的移动. 实际上, 因为地球表面可以用二维空间代表, 我们只需要两个坐标而无须三个. 直观看来, 有人会以南北极(经度)和东西轴(维度)说明, 但也可以从移动中推断出轴. 比方说推销员在伦敦-雅典轴上走的最多, 这个轴可以作为第一个特征值; 第二个特征值与第一个正交. 用这种方式, 推销员在欧洲的每个位置就可以用在这两个特征值上的投影唯一地确定下来. 对它们的投影作图, 就可以显示出旅行路线. 沿着第一个坐标的极端投影对应于雷克雅未克(Reykjavik, 冰岛首都)和莫斯科, 即使它们实际上不在这个轴上.
蛋白质构象也和上面所述的一样. 你看到的极端投影并不一定对应于物理结构, 但是它们可以表征沿轴的运动和总的运动程度. 为了解蛋白质沿构象空间的移动, 我们可以画出特征值2对特征值1的投影图. 为此, 从两个.xvg文件中提取投影数据并且合并到文件ev1-vs-ev2.dat中. 注意'>'表示输出由屏幕重定向到一个文件中, 所以你看不到任何屏幕输出.
为了解释模拟过程, 我们也提取最后7.5 ns(最后1500点)和5.0 ns(最后1000点)的投影, 然后将它们导入xmgrace.
<div class="highlight" style="background:#f8f8f8"><pre style="line-height:125%"><span style="color:#AA22FF">tail</span><span style="color:#666666"> -n </span>1500 ev1-vs-ev2.dat > last-7.5ns.dat <span style="color:#AA22FF">tail</span><span style="color:#666666"> -n </span>1000 ev1-vs-ev2.dat > last-5.0ns.dat <span style="color:#AA22FF">xmgrace</span> ev1-vs-ev2.dat last-7.5ns.dat last-5.0ns.dat </pre></div>投影的形状如何? 它们相互依赖么(分布有所重叠)? 如果只对最后7.5 ns进行分析, 是否得到相同的特征向量(轴)? 使用最后5.0 ns呢?
Write a concluding paragraph, comparing the results obtained for the different proteins. Also reflect on the overall stability and the probability that the structure deposited in the PDB properly reflects the solution structure. In other words, does the structure stay close to the starting structure or does it drift away and how much? Can you suggest a mechanism that would explain the differences in UbcH6 and UbcH8 interaction profiles and why a single conserved mutation can restore the rich interaction profile of UbcH6 when starting from UbcH8? What are the limits of the simulations to explain such a complex biological process?
{kind=link}
{kind=link}