当需要使用AmberTools处理小分子时, 我们首先要安装AmberTools. 在Linux下安装这个工具或许并不太难, 但要想在Windows下安装并不简单. 此外, 如果我们只是使用AmberTools中的几个工具, 将它们全都装上也没有必要. 所以, 我在这里介绍一下在Windows下如何整合一个简单的AmberTools程序包用于处理小分子.
安装Chimera软件后, chimera主目录/bin/amber14目录(根据版本不同, amber后面的两个数字有区别. Chimera 1.10.2中的是amber14, 来源于AmberTools15)就是Chimera自带的CYGWIN下编译的AmberTools, 但可惜的是没有包含所有的程序, 比如拟合RESP电荷的程序就没有包含在其中, 所有我们需要自己编译resp程序.
首先下载AmberTools15.tar.bz2. 解压后, 在AmberTools15主目录/amber14/AmberTools/src/etc中可以找到与resp程序有关的三个源文件resp.F, lapack.F和limits.h. 都是很古老的Fortran源程序, 可以使用安装好的Fortran编译器进行编译. 我使用的是Intel的Fortran编译器2015版, 具体命令是
IntelFortran主目录/ifort.exe /fpp /DCYGWIN resp.F lapack.F
编译选项/fpp用于打开预处理, 因为源代码中包含了预处理命令, 选项/DCYGWIN用于定义宏变量, 使用limits.h中基于CYGWIN的设置. 这两个选项请根据你所用编译器的版本进行适当调整.
编译过程中警告很多, 但都不致命, 最后正常完成, 得到resp.exe. 我们在命令行中试着执行一下resp.exe, 如果输出
usage: resp [-O] -i input -o output -p punch -q qin -t qout -e espot -w qwts -s esout
那就说明我们编译的resp程序可以正常运行了.
如果你还需要将AMBER力场文件转换为GROMACS的拓扑文件, 那你需要acpype.py脚本. 它是一个Python脚本, 为使用它你需要先安装Python 2.x环境, 然后使用Python调用acpype.py, 这不方便, 但我们可以把acpype.py编译成可执行程序acpype.exe, 放在amber14中, 这样发布后就不需要用户安装Python环境了.
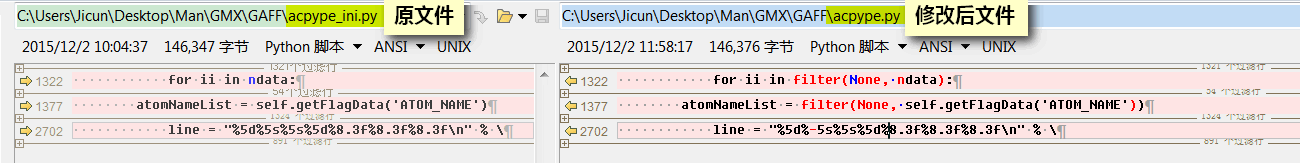
要将Python脚本编译成可执行程序, 我们可以使用py2exe. 首先安装好Python 2.x环境和py2exe, 然后修改acpype.py脚本中的错误
再准备一个setup.py脚本
<table class="highlighttable"><th colspan="2" style="text-align:left">python</th><tr><td><div class="linenodiv" style="background-color: #f0f0f0; padding-right: 10px"><pre style="line-height: 125%"> 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15</pre></div></td><td class="code"><div class="highlight" style="background: #f8f8f8"><pre style="line-height: 125%"><span style="color: #AA22FF; font-weight: bold">from</span> <span style="color: #0000FF; font-weight: bold">distutils.core</span> <span style="color: #AA22FF; font-weight: bold">import</span> setup <span style="color: #AA22FF; font-weight: bold">import</span> <span style="color: #0000FF; font-weight: bold">py2exe</span>
options <span style="color: #666666">=</span> { <span style="color: #BB4444">"py2exe"</span>: { <span style="color: #BB4444">"optimize"</span>: <span style="color: #666666">2</span>, <span style="color: #BB4444">"compressed"</span>: <span style="color: #666666">1</span>, <span style="color: #BB4444">"bundle_files"</span>: <span style="color: #666666">1</span> <span style="color: #008800; font-style: italic"># 此选项只能用于32位版本的Windows</span> } }
setup( options <span style="color: #666666">=</span> options, zipfile<span style="color: #666666">=</span><span style="color: #AA22FF">None</span>, console<span style="color: #666666">=</span>[<span style="color: #BB4444">'</span>acpype<span style="color: #666666">.</span>py<span style="color: #BB4444">'</span>] ) </pre></div> </td></tr></table> 最后执行
python setup.py py2exe
就可以得到一个dist目录, 其中就是acpype.exe及其运行所需的文件.
将我们编译好的resp.exe, acpype.exe及其运行所需的文件全都复制到chimera主目录/bin/amber14/bin目录下, 再将chimera主目录/bin/amber14这个目录一起打包, 就做成了一个在Windows下可以直接使用的AmberTools程序包, 可用于处理小分子. 你如果需要这个工具包, 请点击这里下载. 我整合的这个工具包可用于Windows XP 32位和Windows 7 64位, 其他平台上为测试. 有关此工具包的具体使用方法请参考后续博文.
jhli 抢个沙发。。。Jerkwin 这有啥好抢的. 随时都有的.